


Sr. Editor:
La enfermedad de Kikuchi-Fujimoto (EKF) es una linfadenitis necrosante histiocitaria que se presenta con mayor frecuencia en mujeres jóvenes del sudeste asiático. En España se han publicado 34 casos en los últimos 14 años, de los cuales 15 corresponden a niños.
Sus manifestaciones clínicas son polimorfas, lo cual plantea problemas de diagnóstico diferencial con procesos infecciosos, autoinmunes y/o neoplásicos. La evolución suele ser benigna, si bien se ha descrito su asociación con fenómenos autoinmunes, especialmente con lupus eritematoso sistémico.
Presentamos 2 pacientes con dos extremos del espectro clínico de esta entidad.
Caso 1. Niño de 12 años, que presenta fiebre de 10 días de evolución, y tumoración cervical dura-elástica, sensible a la palpación, de 3 × 4 cm, acompañada de hepatoesplenomegalia, astenia y exantema maculopapuloso. Pruebas complementarias: 4,67 × 109 leucocitos, con 2,87 × 109 neutrófilos. LDH 721 Ul/l. PCR 85 mg/l. VSG 59 mm/h. Tomografía por emisión de positrones (PET): afectación linfática cervical y supraclavicular. TC: conglomerados adenopáticos, con realce periférico y zonas hipodensas centrales (fig. 1). Estudio anatomopatológico: linfadenitis necrosante, compatible con enfermedad de Kikuchi-Fujimoto. Inmunohistoquímica: numerosas células dendríticas plasmocitoides (CD 123, TCL 1), confirmando el diagnóstico.
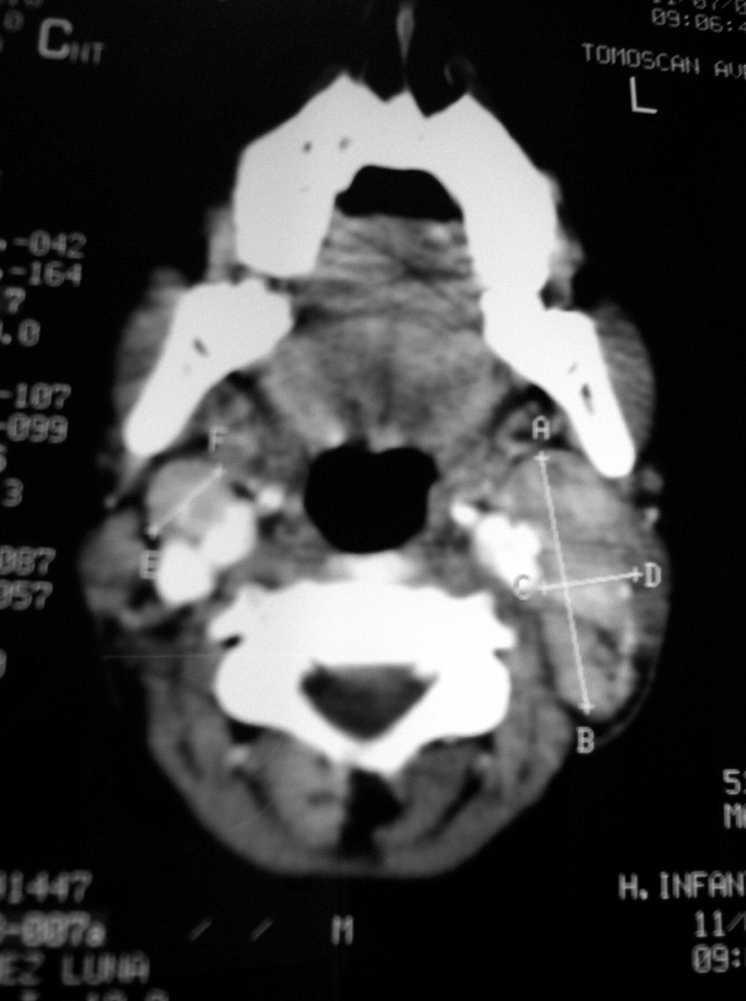
Figura 1.TC. Conglomerados adenopáticos con imágenes sugestivas de necrosis en su interior.
Caso 2. Niño de 2 años que presenta tumoración inguinal de 2 semanas de evolución, dolorosa, hiperémica. 13.000 × 109 leucocitos. Estudio anatomopatológico: linfadenitis necrosante compatible con enfermedad de Kikuchi-Fujimoto (fig. 2). El paciente no presentó ninguna complicación.
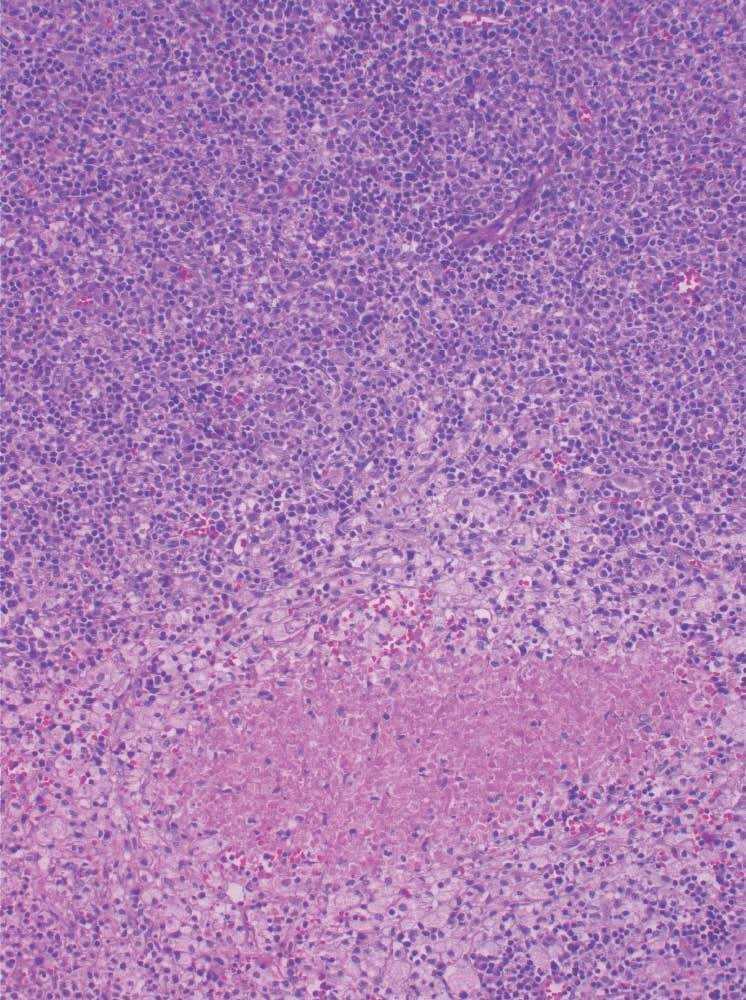
Figura 2. Linfadenitis necrosante. Infiltrado histiocitario (imagen en "cielo estrellado"). (HE × 10.)
Ambos pacientes presentaron un cuadro de linfadenitis aguda. El primero, remedando un cuadro linfomatoso y el segundo, una adenitis inespecífica.
La enfermedad de Kikuchi-Fujimoto es un proceso benigno, autolimitado, descrito por primera vez en 1972 por Kikuchi y Fujimoto. Supone entre un 0,5 y un 5 % de las adenopatías analizadas histológicamente, aunque su incidencia real es desconocida1,7-9.
Etiológicamente se ha relacionado con mecanismos infecciosos e inmunológicos. Esto se basa en el hallazgo en numerosos casos de serologías positivas o presencia de agentes patógenos, tales como: toxoplasma, brucella, yersinia, parvovirus B19, HTLV-1, virus de Epstein-Barr, virus del herpes simple tipo 6, virus de la inmunodeficiencia humana, etc.7,9. No obstante, no ha podido demostrarse que esta sea la causa del proceso, postulándose otras teorías, como la de que podría tratarse de una reacción hiperinmune frente a diversos agentes, entre ellos algunos virus4. También se está revisando la posibilidad de una predisposición genética, pues se observan con más frecuencia algunos HLA de clase II en pacientes con enfermedad de Kikuchi-Fujimoto.
Suele presentarse como fiebre y adenopatías, sobre todo cervicales, dolorosas al tacto y refractarias a tratamiento antibiótico. En algunas de las series más amplias de los últimos años, como son las de Kuo7, y Lin8, se estima que esto ocurre entre un 48 y un 77 % de los casos. En la serie de Lin, Su y Huang9, del año 2005, el 100 % de los pacientes estudiados (23) se presentaron de esta manera, siendo estas adenopatías unilaterales en 19 de ellos (82,6 %), y múltiples en 17 (73,9 %). La fiebre suele ser intermitente y prolongada, con una duración media de alrededor de 20 días1-3, y en algunos casos desaparece tras la biopsia ganglionar, postulándose como causa de este fenómeno la eliminación del foco inflamatorio1,3,7. Otros síntomas asociados son exantema, artralgias, náuseas, vómitos, pérdida de peso y decaimiento10. No obstante, su espectro clínico es muy heterogéneo, lo cual queda ilustrado por nuestros 2 casos, de modo que sólo en el primero la presentación se aproxima clínicamente a la descripción típica de esta enfermedad.
Entre los hallazgos de laboratorio podemos citar la presencia de leucopenia o niveles bajos de leucocitos entre un 67 y un 75 % de los pacientes3,9, no aumento de reactantes de fase aguda, y LDH elevada con transaminasas normales.
El diagnóstico es anatomopatológico, caracterizándose por una linfadenitis necrosante, con ausencia de reacción granulomatosa, y cúmulos histiocitarios alrededor de las áreas de necrosis. Los datos del estudio inmunohistoquímico revelan la presencia de numerosas células dendríticas plasmocitoides.
La evolución es habitualmente benigna, con resolución en pocos meses sin tratamiento específico, lo cual contribuye a su infradiagnóstico. No obstante, existen casos descritos de evolución hacia procesos autoinmunes, como lupus eritematoso sistémico, por lo que es necesario un seguimiento evolutivo5,6.
Esta enfermedad debe ser incluida en el diagnóstico diferencial de linfadenopatías y fiebre junto con otras entidades tales como tuberculosis, infecciones virales, metástasis ganglionares o linfomas, de modo que la presencia de leucopenia y/o zonas de necrosis ganglionar visualizadas mediante técnicas de imagen son orientativas, si bien la confirmación definitiva la dará la anatomía patológica.
Correspondencia: Dra. M.ªJ. Manzano Infante.
Residente de Pediatría.
Olivar del Loreto, 26. 41807 Espartinas. Sevilla. España.
Correo electrónico: mjmanzano@msn.com
