

La lectina de unión a la manosa (mannose-binding lectin [MBL]) es una proteína sérica perteneciente al sistema inmunitario innato. Se une a los azúcares de las membranas de múltiples microorganismos, favoreciendo su opsonización y eliminación. El déficit de MBL resulta del polimorfismo del gen MBL2 y se asocia a una amplia variedad de infecciones recurrentes, incluidas las infecciones del tracto respiratorio. Presentamos un caso de displasia ectodérmica anhidrótica asociada a un déficit de MBL, inmunodeficiencia nunca descrita en pacientes afectados de displasia ectodérmica anhidrótica.
Mannose-binding lectin (MBL) is a serum protein of the innate immune system. MBL enhances opsonophagocytosis by binding to carbohydrates expressed by multiple pathogens. MBL deficiency is due to polymorphisms in the structural and promoter sequences of the MBL2 gene and is associated with variety of recurrent infections, including respiratory tract infections. We present a case of anhidrotic ectodermal dysplasia associated with severe mannose-binding lectin deficiency, never described in patients with anhidrotic ectodermal dysplasia.
La displasia ectodérmica (DE) comprende un amplio y heterogéneo grupo de enfermedades genéticas no progresivas ocasionadas por defectos primarios en el desarrollo de 2 o más tejidos derivados del ectodermo embrionario1-8. Su fenotipo es complejo, asociando disfunción de glándulas sudoríparas, defectos en dientes, pelos y uñas, lesiones cutáneas y dismorfias craneofaciales6,8. Es una enfermedad rara, con una incidencia de 1/10.000 o 1/100.000 recién nacidos vivos, siendo más frecuente en varones5,8. A nivel molecular se produce mutaciones en el gen de la ectodisplasina (EDA), del receptor de la EDA (EDAR) o del receptor de la EDA asociado al dominio de muerte (EDARADD)3,6,9. El gen EDA está mutado en las displasias ectodérmicas anhidróticas (DEA) ligadas al cromosoma X (EDA; MIM 305100), constituyendo la variante más común3,6,7,9. EDAR y EDARADD son los genes mutados tanto en las formas autosómicas dominantes (MIM129490) como en las recesivas (MIM 224900)3,6,7,9. Los casos ligados al cromosoma X pueden asociarse a inmunodeficiencias10, por lo que es frecuente que asocien además infecciones recurrentes presentes en solo un tercio de los pacientes con DEA con herencia autosómica11. Presentamos un caso de DEA asociado a un déficit severo de MBL y a un trastorno de la β-oxidación.
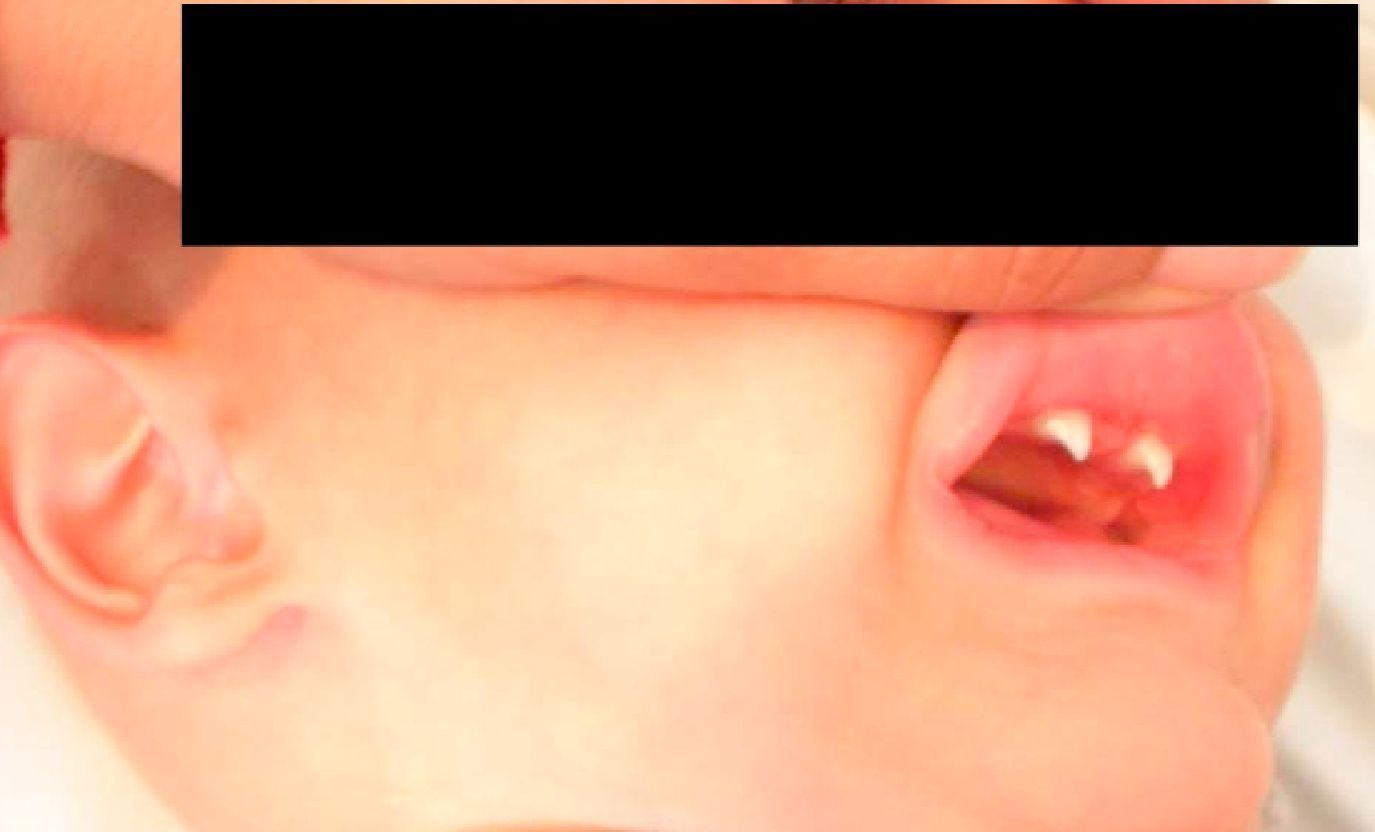
Caso clínicoVarón de 3 meses y medio de vida seguido en consultas de nutrición y neumología de nuestro centro por trastorno de la β-oxidación de ácidos grasos de cadena media, diagnosticado presintomáticamente mediante cribado neonatal ampliado, junto a fallo de medro, desnutrición y múltiples ingresos hospitalarios por neumonías de probable etiología vírica (hemograma con linfocitosis y PCR baja) y bronquitis recurrentes con mala evolución pese al tratamiento preventivo con budesonida (200μg/12h) y, una vez alcanzados los 6 meses de edad, con montelukast (4mg/24h). Como antecedentes familiares de interés destacan etnia gitana y trastornos en la dentición materna desde la infancia. Padres no consanguíneos. Exploración física: longitud de 71cm (p25-50), peso de 6,1 kg (<p3). Ligera palidez mucocutánea, fontanela anterior amplia (2,5×2,5cm) y normotensa, pelo ralo, débil, pajizo y raíz nasal ancha, escaso panículo adiposo y llanto disfónico. Frecuencia cardíaca: 110lpm. Frecuencia respiratoria: 45rpm. Saturación de oxígeno del 96%, sin oxigenoterapia suplementaria. Tiraje subcostal e intercostal. A la auscultación cardiopulmonar hallamos crepitantes diseminados en ambos campos pulmonares y espiración alargada, sin soplos cardíacos. El resto de la exploración, sin hallazgos patológicos. Ante estos datos clínicos se realizan diversas pruebas complementarias, con los siguientes resultados: hemograma: leucocitos 18,17×109/l (58,1%N, 27%L, 12,9%M), Hb 86g/l, VCM 72,8 fL, HCM 24,4pg, Hcto. 0,257 l/l, plaquetas 636×109/l; alfa-1-antitripsina: 179mg/dl; Mantoux: 0mm; varios tests del sudor imposibles de realizar por ausencia de sudoración; complemento, inmunoglobulinas y subpoblaciones linfocitarias, normales para su edad. Los niveles de MBL cuantificada mediante técnica ELISA, (BIOPORTO, Gentofte, Dinamarca) son de 93,27ng/ml; test yodo-almidón: ausencia total de sudoración. TACAR pulmonar (fig. 1): imagen de condensación en la base pulmonar derecha. Tras los hallazgos clínicos, radiológicos y analíticos descritos, se establece el diagnóstico de DEA. Presenta evolución posterior favorable, tras realizar tratamiento con salmeterol/fluticasona 25/50 (2 puf/12h) y azitromicina (10mg/kg/24h, 3 días cada 15 días), encontrándose 2 años más tarde asintomático desde el punto de vista respiratorio, sin tratamiento preventivo, con buena ganancia ponderal y con aparición de incisivos cónicos (fig. 2). Tras intentar nuevamente sin éxito el test del sudor, se ha realizado un estudio genético de fibrosis quística que ha resultado negativo. La secuenciación del gen NEMO (NF-κB essential modulator, asociado con un defecto de los receptores toll-like) no mostró alteraciones del sistema inmunitario innato.
Discusión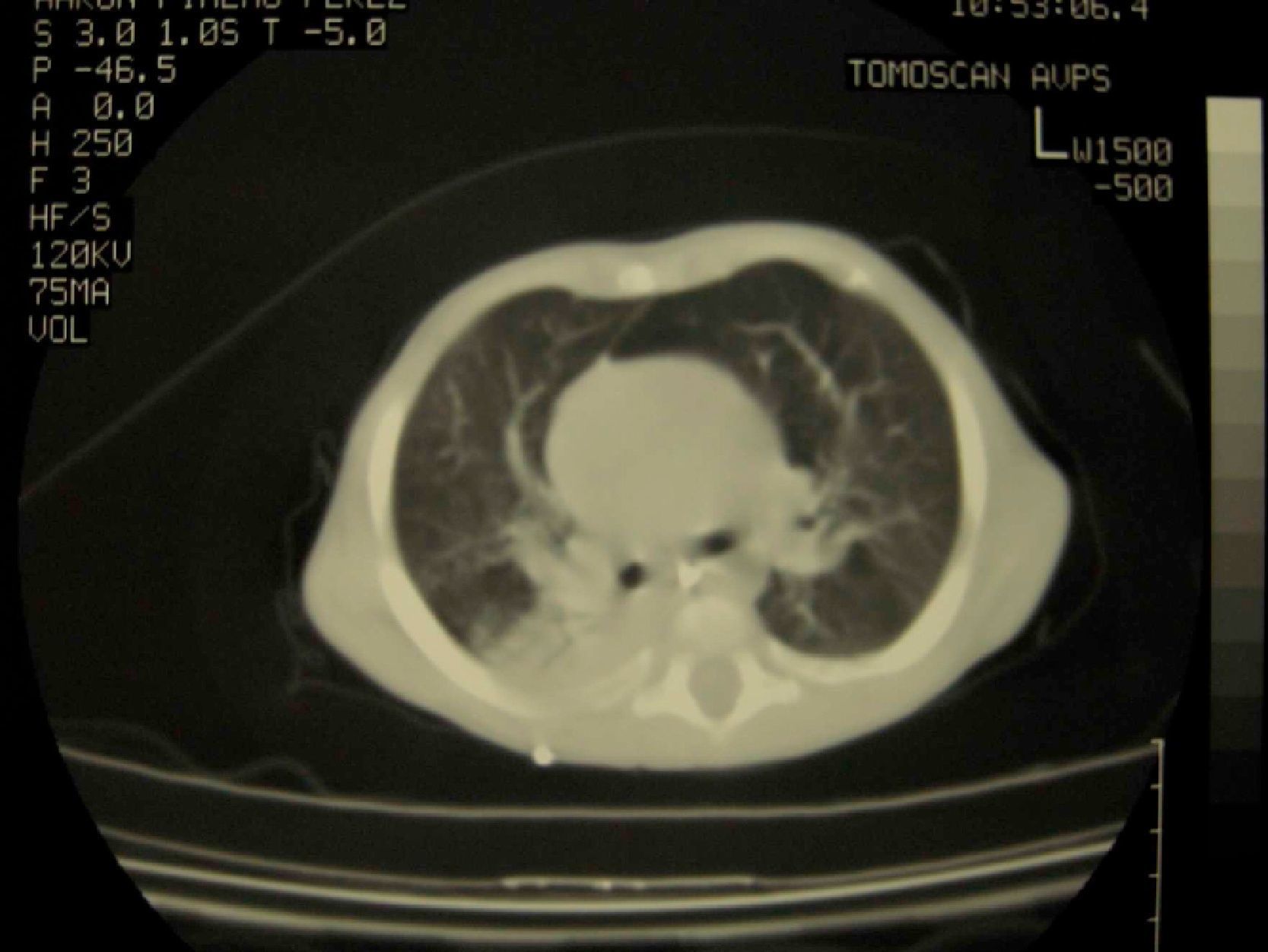
La MBL es una proteína sérica perteneciente a la familia de las colectinas, con una estructura similar al C1q11. Ambas, tanto MBL como C1q, son proteínas del sistema immunitario innato que pueden activar el complemento a través de las proteasas asociadas12-14. El C1q activa el complemento por la vía clásica y la MBL inicia la vía de las lectinas del complemento después de su unión a los azúcares de la estructura de múltiples microorganismo, incluidos patógenos respiratorios tales como virus influenza A, Haemophilus influenzae, Mycoplasma pneumoniae, complejo Mycobacterium avium, Legionella pneumophila, Nocardia farcinica o coronavirus, Pseudomonas aeruginosa, Burkholderia cepacia y Pneumococcus12,13,15.
El déficit de MBL se asocia a una amplia gama de fenotipos infecciosos, incluyendo enfermedad neumocócica invasiva, sepsis grave, sepsis meningocócica, infecciones graves tras quimioterapia y trasplante alogénico de células madre hematopoyéticas, duración prolongada de fiebre neutropénica en pacientes oncológicos e infecciones del tracto respiratorio12,13,16.
Hay 2 genes humanos de MBL: MBL1, que es un seudogén, y gen MBL2, que codifica para la proteína MBL14. Existe una significativa variación en los niveles séricos de MBL de una persona a otra, como resultado del polimorfismo del gen MBL213,14,16 existiendo 3 polimorfismos estructurales y 3 del promotor12-14,16. El polimorfismo estructural consiste en polimorfismos de los codones 52 (CGT3TGT), 54 (GGC3GAC) y 57 (GGA3GAA)12,14,16. A los alelos mutantes de dichos codones se les denomina D, B y C, respectivamente12,14,16. Una región de codificación que contenga cualquiera de las mutaciones D, B o C se conoce como O y el alelo de tipo salvaje en cada lugar se conoce como A14–16. Las mutaciones estructurales impiden la formación de la triple hélice en la cola de la MBL colágeno, lo que lleva a la degradación y la deficiencia funcional de la MBL13. Las concentraciones séricas de MBL son un 10–20% de lo esperado en los heterocigotos (A/O) para los polimorfismos estructurales y prácticamente indetectables en los homocigotos funcionales (O/O, los individuos que son homocigotos o heterocigotos compuestos para dos mutaciones diferentes de codificación)15–16. Los polimorfismos en la región promotora del gen MBL2 también ejercen efectos funcionales en la transcripción de MBL16.
La MBL humana recombinante que tiene la misma función en la activación del complemento ha sido obtenida derivada del plasma y utilizada en ensayos clínicos en fases iniciales en la quimioterapia y ajustes postrasplante del trasplante de hígado13. Si cada vez son más numerosos los datos de la función de MBL en la patogénesis de la infecciones, sería razonable considerar esto como una condición de objetivo atractivo para el reemplazo de MBL, dada su alta prevalencia y mortalidad13. La evidencia de una disminución en la supervivencia de los pacientes con FQ con genotipos MBL2 variante sugiere que estos pacientes podrían beneficiarse de la suplementación intermitente con MBL13. In vitro se ha descrito que la administración de la MBL derivada del plasma (1.000-5.000ng/ml) en pacientes con déficit de MBL produce una restauración de la función de opsonización12. Por lo tanto, los pacientes con déficit de MBL podrían beneficiarse de la terapia de sustitución con MBL12. Existe un ensayo en fase I donde la administración de MBL parece segura y los niveles séricos aumentan hasta niveles normales, pero su vida media presentó variabilidad12. Algunos pacientes con infecciones recurrentes muestran mejoría clínica después de la terapia de remplazo de MBL12. El manejo de estos pacientes debe incluir además un tratamiento intensivo de las infecciones intercurrentes, una correcta vacunación antigripal, anti-Haemophilus y antineumocócica, e incluso pueden beneficiarse también del uso de antibioterapia profiláctica, tal y como hicimos en nuestro paciente.
Varios estudios sugieren que el déficit de MBL, como parte del sistema inmunitario innato, desempeña un papel más importante en los primeros meses de vida, especialmente entre 6-17 meses de edad, cuando se está desarrollando su sistema inmunitario adaptativo («ventana de vulnerabilidad»)17,18. Esto podría justificar una posible mejoría clínica espontánea en nuestro paciente, atribuible a una mayor edad del paciente y a un mayor desarrollo del sistema inmunitario adquirido. No hemos encontrado asociación específica entre defectos de β-oxidación e inmunodeficiencias. En las metabolopatías pueden producirse inmunodeficiencias secundarias por efecto tóxico y daño mitocondrial. Existen síndromes específicos que asocian inmunodeficiencia y DEA debido a mutaciones en genes que afectan a los factores de transcripción nuclear implicados en la señalización intracelular de TLRS: DEA con síndrome hiper-IgM (X-linked) secundaria a mutaciones en el gen NEMO y que produce disfunción de células T y B19 y DEA con inmunodeficiencias de células T (autosómica dominante y ligada al cromosoma X) por mutaciones en NFKBIA20.
Como conclusión, es necesario afirmar que debemos descartar inmunodeficiencias, incluido el déficit de MBL en pacientes con DEA, especialmente si asocia infecciones respiratorias de repetición. La terapia sustitutiva con MBL se plantea como una posible futura arma terapéutica en los pacientes con déficit de MBL. La asociación del déficit de MBL podría potenciar la gravedad de las manifestaciones clínicas en los pacientes con DEA, tal y como ocurre en otras inmunodeficiencias15. Considerando que el déficit severo de MBL tiene una incidencia del 10% en la población general, la asociación entre déficit de MBL y DEA en nuestro paciente podría deberse al azar, por lo que son necesarios más estudios que evalúen esta observación.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
