



La epilepsia parcial continua es una forma de estatus epiléptico parcial, que se caracteriza por la presencia de mioclonías repetidas que afectan a un grupo muscular. Su origen es cortical y pueden prolongarse durante horas, días, semanas y excepcionalmente años. Dentro de estas formas de epilepsia podemos diferenciar 2 grupos: el primer grupo o síndrome de Kojewnikow clásico, comprende a niños con una lesión conocida en la región rolándica (cuya etiología es también conocida) y existe un daño neurológico estable (salvo si la lesión aumenta, como por ejemplo, los tumores). Consiste en la presencia de crisis parciales motoras, a veces seguidas por períodos de mioclonías bien localizadas. El segundo grupo o síndrome de Rasmussen se caracteriza por inicio de crisis en pacientes previamente sanos, comenzando con crisis parciales motoras a las cuales rápidamente se asocian mioclonías que pueden afectar distintas zonas corporales. La evolución es progresiva, con deterioro neurológico.
Describimos el caso de un niño de 7 años de edad estudiado por crisis convulsivas parciales y degeneración progresiva de funciones superiores. Se le practican estudios de imagen, neurofisiológicos y pruebas de laboratorio, siendo diagnosticado de síndrome de Rasmussen. Finalmente, se le realiza una hemisferectomía paliativa, confirmándose el diagnóstico de encefalitis de Rasmussen mediante biopsia.
Continuous partial epilepsy is a form of partial status epilepticus, which is characterized by the presence of repeated myoclonus affecting a muscle group. Its origin is cortical and it can last for hours, days, weeks and exceptionally, years. Within these forms of epilepsy we can distinguish two groups: the first group or Kojewnikow classic syndrome includes children with a known lesion in the rolandic region (the etiology is also known) and there is a stable neurological damage (unless the injury increases, e.g., tumors). This disease is characterized by the presence of motor partial seizures, sometimes they are followed by periods of well-localized myoclonus. The second group or Rasmussen syndrome is characterized by onset of seizures in previously healthy patients, starting with partial motor seizures, that later can be combined with myoclonus that affect different areas of the body. It is a progressive disease that leads to neurological damage.
A case is presented of a 7-year-old patient investigated due to having partial seizures and progressive neurological degeneration. After performing imaging studies, neuropsychological studies, and laboratory tests, he was diagnosed with Rasmussen's syndrome. Finally, a palliative hemispherectomy was performed and the diagnosis was confirmed by a biopsy.
La epilepsia parcial continua1 es una forma de estatus epiléptico parcial, que se caracteriza por la presencia de mioclonías repetidas que afectan a un grupo muscular. Su origen es cortical y pueden prolongarse durante horas, días, semanas y excepcionalmente años. Dentro de estas formas de epilepsia podemos diferenciar 2 grupos2: el primer grupo o síndrome de Kojewnikow clásico, comprende a niños con una lesión conocida en la región rolándica, de etiología también conocida. Existe un daño neurológico estable, salvo que se trate de una lesión progresiva, como un tumor. La clínica consiste en presencia de crisis parciales motoras, que pueden ser seguidas por períodos de mioclonías bien localizadas. Las pruebas de imagen suelen mostrar la lesión cerebral y el EEG revela alteraciones localizadas con trazado de fondo normal, sobre todo a nivel rolándico o central. Si la lesión es sugestiva de tratamiento quirúrgico, la clínica generalmente desaparece tras la extirpación de la misma. El segundo grupo o síndrome de Rasmussen se caracteriza por inicio de crisis en pacientes previamente sanos, comenzando con crisis parciales motoras a las cuales rápidamente se asocian mioclonías, que pueden afectar a distintas zonas corporales. La resonancia magnética (RM) suele mostrar atrofia en el hemisferio afectado, especialmente en fases avanzadas de la enfermedad. El EEG revela un trazado de fondo anormal con alteraciones paroxísticas focales o generalizadas. Se trata de una entidad progresiva, presentando a lo largo de su evolución deterioro neurológico. El hallazgo de lesiones inflamatorias en las biopsias cerebrales sugiere que este tipo de epilepsia sería el resultado de una encefalitis crónica. Responde mal al tratamiento anticomicial y la hemisferectomía, pese a sus importantes efectos secundarios, continúa siendo el único tratamiento que ha demostrado ser capaz de frenar el avance de la enfermedad y la aparición de crisis en la mayoría de los casos.
Caso clínicoSe presenta el caso de un niño de 7 años que ingresa en nuestro hospital para estudio por crisis convulsivas parciales y alteración de las funciones superiores. Entre sus antecedentes familiares no existe consanguinidad, ni abortos ni muertes en el período neonatal. Su padre presenta problemas de salud mental de tipo ansioso y su madre refiere dislexia en edad escolar. Tiene una hermana de 14 años con problemas de aprendizaje. No constan en su historia antecedentes perinatales de interés y se trata de un niño previamente sano, con un desarrollo psicomotor, hasta el inicio de los síntomas, apropiado. Destaca que desde los 6 años de edad, aproximadamente 1 año antes de su ingreso, comienza a manifestar episodios nocturnos de somniloquia y «body-rocking», sin un desencadenante previo, y desde los 6 años y medio de edad, asocia deterioro cognitivo lentamente progresivo con disminución del rendimiento escolar, alteraciones de la memoria y temblor en miembro superior derecho.
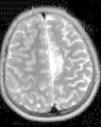
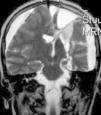
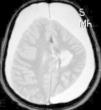
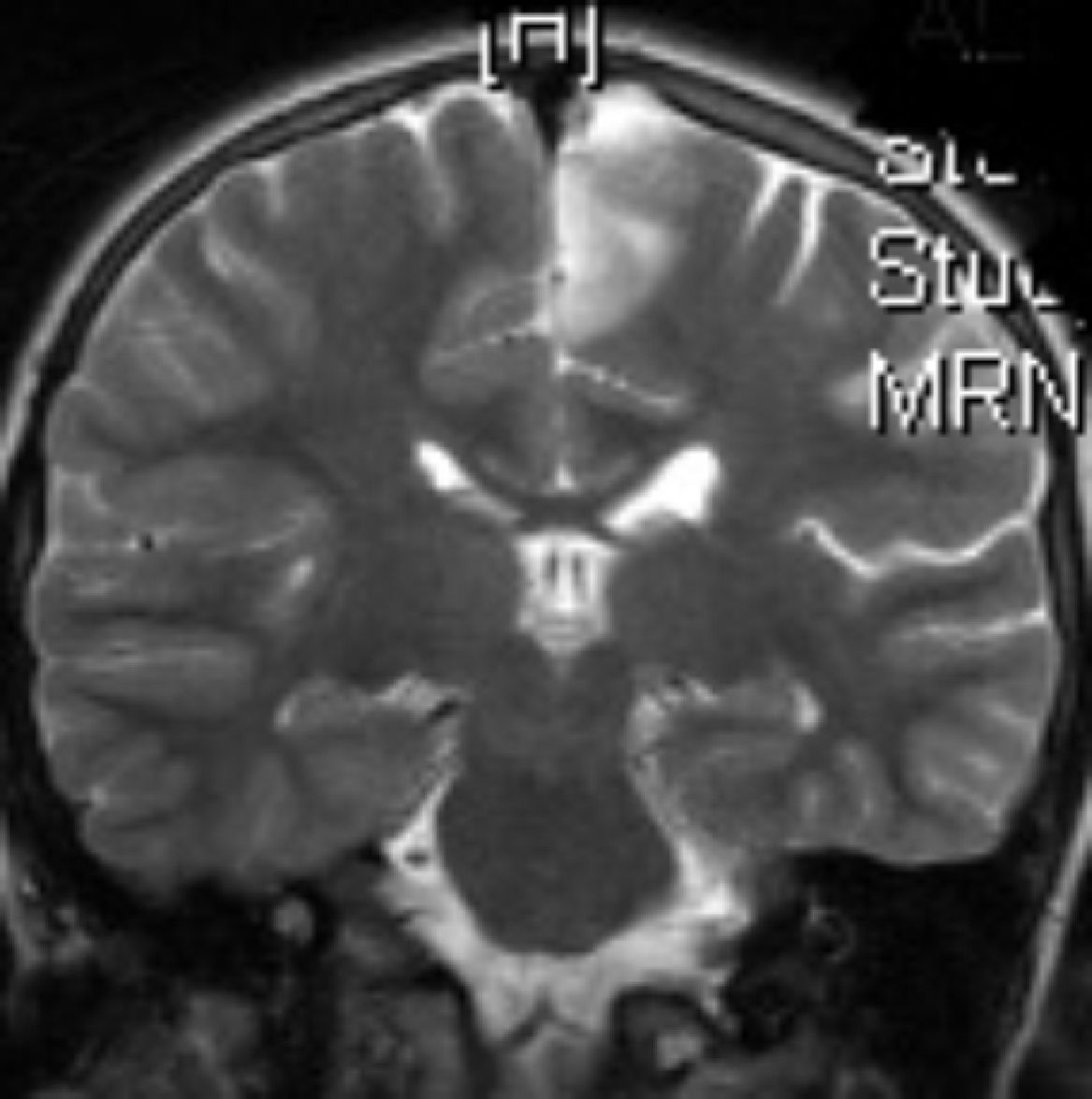
Ingresa en nuestro centro a los 7 años de edad, por presentar varias crisis tónico-clónicas que afectan principalmente al hemicuerpo derecho, con pérdida de fuerza posterior a este nivel. En el TC craneal al ingreso se observaron lesiones hipodensas a nivel parafalcial anterior izquierdo y fronto-parietal izquierdo y en la RM cerebral lesiones córtico-subcorticales a nivel frontal superior, frontal medio y parietal, de aspecto tumefactivo y atrofia marcada del hemisferio cerebral izquierdo (figs. 1 y 2). No se apreciaron alteraciones en el árbol vascular. En el EEG presentaba asimetría interhemisférica con gran lentificación en el hemisferio izquierdo y actividad paroxística focal con ondas agudas, puntas y complejos punta-onda de elevado voltaje, principalmente a niveles frontal y fronto-temporal izquierdos. La ecografía Doppler de troncos supraórticos no mostró alteraciones. Se solicitaron estudios de laboratorio, incluyendo estudio metabólico completo y serologías virales, todos ellos anodinos. La citoquímica de líquido cefalorraquídeo (LCR) fue normal y las PCR de virus neurotropos en LCR negativas. Tampoco se apreciaron células malignas. Otros estudios de inmunología realizados comprenden anticuerpos onconeuronales, negativos; ANA 1/80 patrón moteado, y anti-RNP-A positivo. Como pruebas de segundo nivel solicitamos RM cerebral con espectroscopia que mostró varias lesiones córtico-subcorticales, alteraciones en el cociente N-acetilaspartato/colina e inversión en el cociente de lípidos/lactato, sin demostrarse aumento de lactato en el LCR. Otros estudios llevados a cabo: fondo de ojo, estudio cardiológico, audición, biopsia de piel y de músculo, resultaron normales. Los potenciales evocados visuales no resultaron alterados y en los potenciales evocados somatosensoriales se encontraron anomalías en la vía somatosensorial procedente de ambas extremidades superiores, con respuesta cortical de amplitud reducida bilateralmente. Bajo la sospecha de síndrome de Rasmussen se envía a hospital de referencia, donde realizaron nueva RM cerebral (secuencias DWI, T1, DP-T2, FLAIR, T1D3) con aumento de surcos corticales y pérdida de volumen del hemisferio cerebral izquierdo, compatible con esta enfermedad.
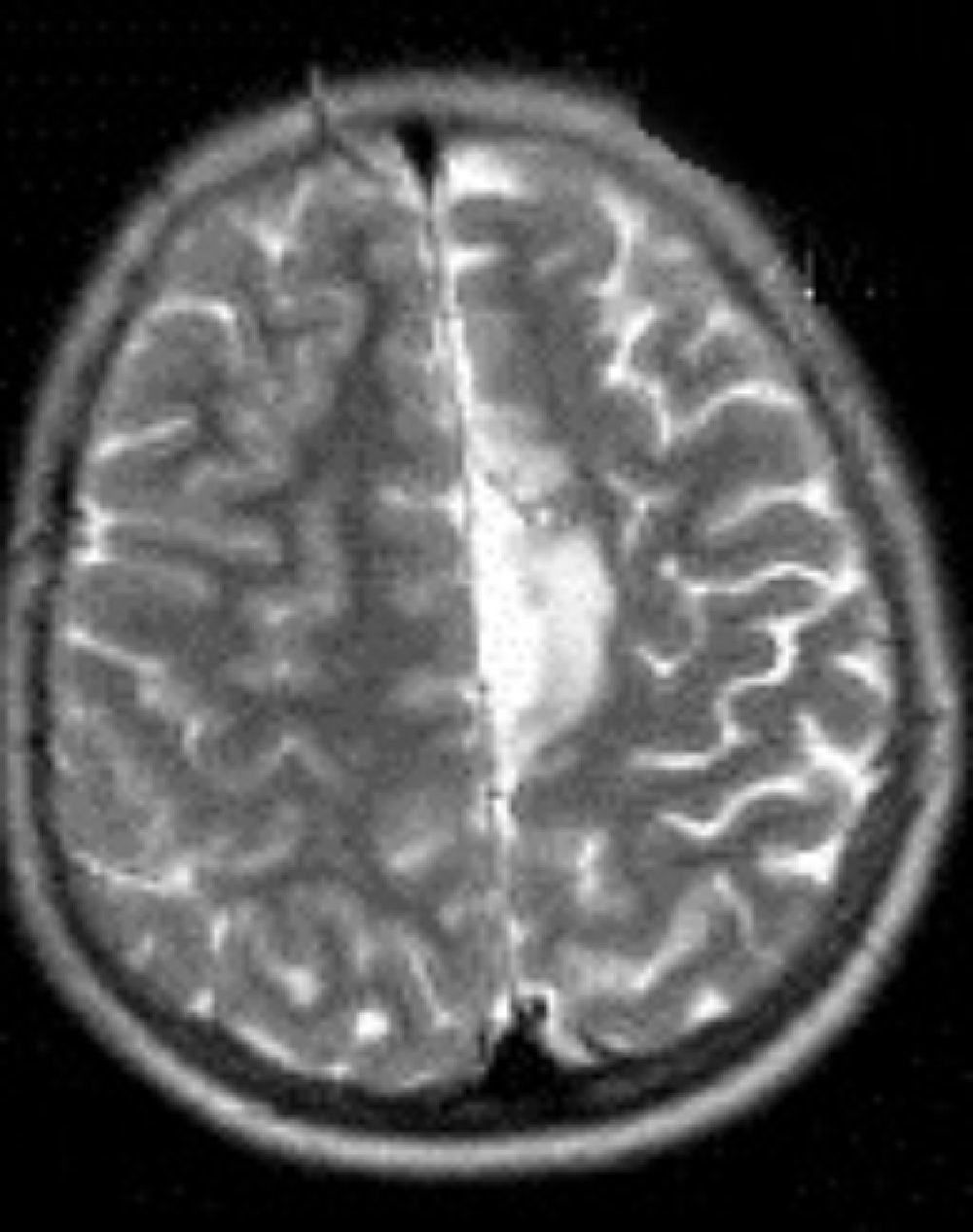
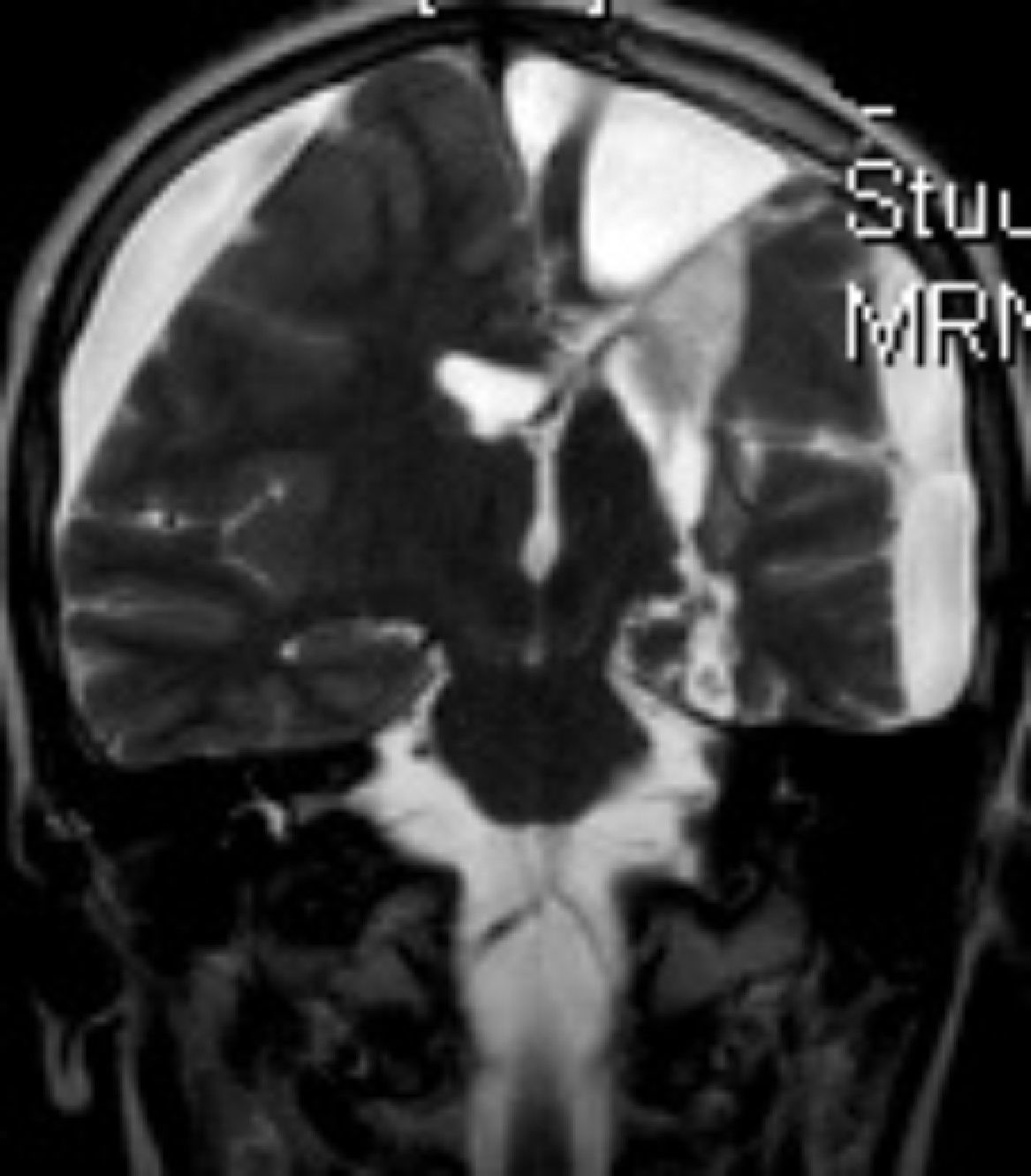
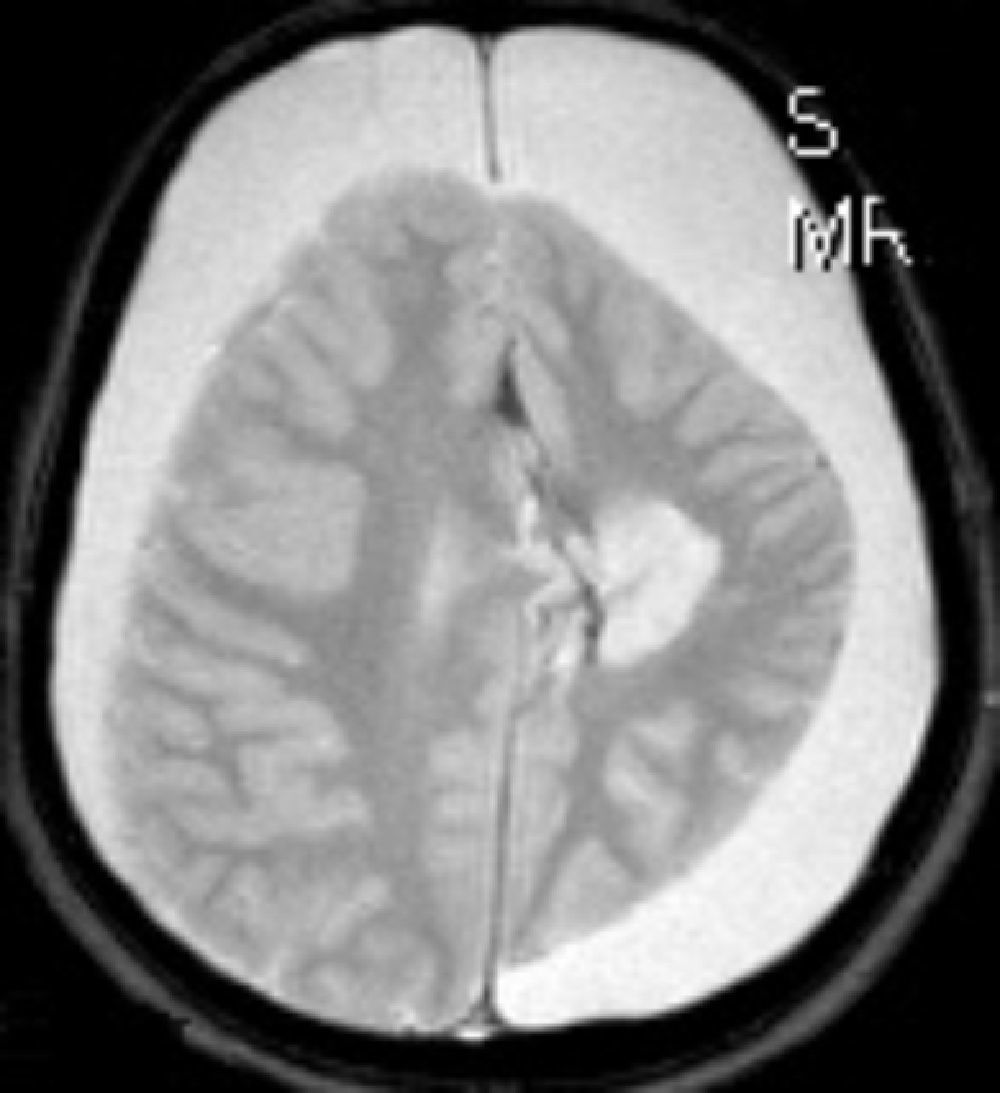
En cuanto a las medidas terapéuticas, inicialmente recibió tratamiento con carbamacepina, con empeoramiento del cuadro (aparición de mioclonías frecuentes en extremidades derechas). Se interrumpió el tratamiento y se instauraron clonazepam y clobazam, con mejoría a corto plazo. En los meses siguientes presentó aumento de mioclonías en los miembros derechos, pérdida de fuerza mantenida en los mismos y aparición de algún episodio de crisis parciales y alguna convulsión tónico-clónica generalizada. Ante el empeoramiento, se añadieron al tratamiento ácido valproico, corticoides y recibió 4 ciclos de inmunoglobulinas. No se volvieron a registrar convulsiones tónico-clónicas, pero se incrementó la frecuencia de crisis parciales y mioclonías. La degeneración motora y cognitiva también progresó, siendo incapaz de pronunciar muchas palabras, persistiendo paresia del miembro superior derecho y dificultades para la deambulación. Dado el fracaso del tratamiento médico, se decide remitir al mismo hospital de referencia para realización de tratamiento quirúrgico paliativo. A los 8 años y 8 meses de edad se le practica una hemisferectomía izquierda por vía frontal (figs. 3 y 4). En el acto quirúrgico se obtuvo una biopsia cerebral con inflamación perivascular, gliosis y degeneración neuronal compatible con encefalitis de Rasmussen.
Actualmente, el niño tiene 10 años de edad y es seguido en consultas de Neurología y consultas de Rehabilitación de nuestro centro. No ha presentado crisis comiciales desde su intervención. Presenta hemiplejía derecha, fuerza conservada en extremidades izquierdas y control del tronco en sedestación. Se encuentra alerta, pero desorientado y con lentitud de ejecución; entiende frases sencillas y responde con gestos. Es capaz de deglutir, no controla la micción y sí la defecación. Se mantiene actualmente tratamiento con ácido valproico, fenobarbital, metilfenidato y mirtazapina.
DiscusiónEl síndrome de Rasmussen es una enfermedad neurológica rara. Su edad de inicio es entre los 14 meses y los 14 años3, con una media alrededor de los 7 años de edad. La etiología se desconoce, aunque se han formulado varias hipótesis al respecto. Inicialmente, se pensó en una causa viral de la encefalitis, puesto que las células T que forman parte de los infiltrados inflamatorios expresan complejos de antígenos de histocompatibilidad mayor (CHCM) similares a los expresados en caso de pacientes afectados por encefalitis virales. Power et al.4 y Walter et al.5 en sendos estudios aíslan material genético de citomegalovirus y virus de Epstein-Barr, respectivamente, en pacientes diagnosticados de síndrome de Rasmussen. La teoría más aceptada actualmente es la autoinmunitaria. En un estudio llevado a cabo por Whitney et al.6 detectaron anticuerpos contra los receptores de glutamato (subunidad GluR3) capaces de activar la vía clásica del complemento, produciéndose citólisis neuronal. Apoya esta teoría el hecho de que muchos pacientes mejoran, al menos transitoriamente, tras la plasmaférisis. Sin embargo, otros autores consideran estos autoanticuerpos como marcadores de epilepsias refractarias en general7. Las teorías más recientes intentan explicar la etiopatogenia de esta enfermedad como resultado de una alteración de la barrera hematoencefálica por causas desconocidas, lo cual propiciaría el paso de autoanticuerpos al encéfalo. Estos serían los responsables tanto de la inflamación y daño local como de la hiperexcitabilidad que daría lugar a las crisis epilépticas8. Atkins et al.9 sugiere que podría existir una cierta predisposición genética, con un primer desencadenante de la enfermedad de origen viral, siendo mantenida la encefalitis mediante un mecanismo autoinmunitario. En nuestro caso no se conocen antecedentes de una encefalitis viral y las serologías virales en LCR fueron negativas.
El síndrome de Rasmussen se manifiesta clínicamente como una epilepsia parcial continua, desarrollándose finalmente una epilepsia refractaria10. Podemos distinguir 2 subtipos: el predominante, que afecta a la corteza de un hemisferio cerebral, manifestándose como crisis convulsivas focales (pueden generalizarse siendo característicamente unilaterales) y mioclonías, y el segundo subtipo, con afectación de los ganglios de la base, presentándose como movimientos paroxísticos anormales, tales como distonías. A lo largo de la enfermedad, se añaden otros síntomas11, como hemiparesia, hemiplejía, hemianopsia y degeneración en las funciones superiores. Ello se intenta explicar debido a una gran pérdida de neuronas secundaria a las crisis comiciales frecuentes. Nuestro paciente comenzó presentado crisis epilépticas parciales que afectaban a los miembros derechos con pérdida de fuerza posterior, apareciendo más tarde mioclonías. Presentó también afectación de las funciones superiores, inicialmente alteraciones de memoria y progresivamente disartria y afasia.
El diagnóstico de síndrome de Rasmussen3 se basa en una clínica compatible, junto con estudios de imagen con la afectación característica, descartadas otras enfermedades. La prueba definitiva sería la biopsia cerebral, pero esta no es recomendable realizarla salvo a partir de la materia cerebral extraída en la hemisferectomía. Son hallazgos característicos la presencia de actividad lenta sobre un hemisferio en el electroencefalograma, interrumpida por puntas multifocales. La RM es la técnica de imagen que más información aporta, siendo característica la observación de tumefacción de las zonas más dañadas y de atrofia del hemisferio afectado. También son de gran utilidad la espectroscopía por RM, la PET y la SPECT. En la primera podemos observar alteraciones en los patrones de colina y creatinina (marcadores de inflamación cerebral); la segunda es útil como marcador de la actividad de la microglía y la última, permite visualizar incrementos del flujo cerebral en las áreas afectadas durante las crisis convulsivas. El estudio del LCR muestra bandas oligoclonales en algunos pacientes. Por último, en la biopsia cerebral pueden observarse hallazgos compatibles con encefalitis, tales como infiltración perivascular de células redondas, nódulos de microglía, neuronofagia y degeneración espongiforme12. En nuestro caso las pruebas de imagen, laboratorio y EEG apuntaban hacia el síndrome de Rasmussen, hasta que finalmente, la biopsia cerebral lo confirmó.
En lo que se refiere al tratamiento de esta patología, se han intentado controlar las crisis con combinación de fármacos anticomiciales y esteroides, sin resultados satisfactorios. En algunos casos se ha observado remisión durante cierto tiempo con inmunomoduladores como la ciclofosfamida y la administración de inmunoglobulina por vía intravenosa13 (dosis inicial de 400mg/kg 5 días y dosis de mantenimiento de 0,4mg/kg/día) proporciona mejoría y control de las crisis en algunos pacientes. La plasmaféresis14 muestra buenos resultados inicialmente pero a largo plazo pierde efectividad. El tratamiento clásico es la hemisferectomía15, que continúa siendo el único que ha demostrado frenar el avance de la enfermedad y la aparición de crisis hasta en el 90% de los casos. En nuestro caso el tratamiento con anticomiciales y corticoides, no resultó efectivo. Nuestro paciente recibió también 4 ciclos de inmunoglobulinas por vía intravenosa, aunque no se volvieron a registrar convulsiones tónico-clónicas, las crisis parciales persistieron con frecuencia elevada y la degeneración motora y cognitiva continuó su curso. La hemisferectomía izquierda permitió el control de las crisis epilépticas, mejorando la calidad de vida del niño y permitiendo el avance en su rehabilitación; presenta como consecuencia hemiplejía derecha, entre otras graves secuelas.
En resumen, es necesario pensar en esta patología cuando nos encontremos frente a un caso de epilepsia parcial continua junto con hemiparesia en el lado afecto por las crisis y que responde mal a fármacos anticomiciales. La clínica compatible junto con las pruebas de imagen sugestivas de esta enfermedad, especialmente RM y electroencefalograma pueden ser suficientes para su diagnóstico. Pese a los avances en el tratamiento médico, la hemisferectomía continúa siendo el tratamiento más eficaz, que aunque presenta múltiples efectos secundarios, puede controlar las crisis, mejorando la calidad de vida del paciente.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
