
Sr. Editor:
El déficit de acil-coenzima A deshidrogenasa de cadena media (MCAD) es el trastorno más frecuente de la oxidación de los ácidos grasos. Diversos estudios señalan que éste y otros defectos enzimáticos del metabolismo de los ácidos grasos son responsables de casos atribuidos a síndrome de muerte súbita del lactante y síndrome de Reye precoz.
Lactante de etnia gitana de 6 meses acude a urgencias por somnolencia e hiporreactividad en las últimas 6h. Padecía síntomas catarrales con fiebre de hasta 39,2°C axilar desde hacía 2 días, sin ingesta de alimentos en las 12h previas. Los padres eran primos hermanos y, en la exploración física, destacaba una faringe hiperémica con mucosidad abundante, subcrepitantes bibasales, decaimiento y somnolencia con buena respuesta a estímulos dolorosos y sin signos de focalidad. Presentaba un desarrollo ponderoestatural adecuado, genitales normoconfigurados y ausencia de defectos en la línea media. En las analíticas realizadas se observó discreta leucocitosis con linfocitosis, glucemia 12mg/dl, GOT 68 UI/l, urea 65mg/dl, proteína C reactiva (PCR) 1,28g/dl y cuerpos cetónicos urinarios. Se realizaron, además, pruebas de creatinfosfoquinasa (CPK), ácido láctico, amonio, equilibrio ácido-base, bioquímica y citología en el líquido cefalorraquídeo (LCR), radiografía de tórax, cultivos de sangre, heces, orina y LCR, con resultaron negativos. Ingresó tras reposición intravenosa de glucosa, y posteriormente se realizaron ecografías transfontanelar y abdominal, electroencefalograma (EEG) y determinaciones plasmáticas en el momento de la hipoglucemia de hormona de crecimiento, cortisol, insulina/glucemia, hormona tiroestimulante (TSH) y T4 libre sin hallazgos patológicos. Se estudiaron aminoácidos y ácidos orgánicos en plasma, orina y LCR, con hallazgos compatibles con déficit de MCAD (valores supranormales de acilglicinas en orina y de acilcarnitinas de 6 a 8 átomos de carbono en plasma, con carnitina libre, total y esterificada normales). Se inició tratamiento con carnitina, riboflavina y medidas dietéticas. El estudio genético confirmó la condición de homocigoto de la paciente para la mutación G985A en el gen de la MCAD, y los padres y 2 de sus 3 hermanos resultaron portadores de la misma. Los resultados del estudio familiar (tabla 1) muestran una excelente correlación genético-bioquímica, hallazgo bien conocido1.
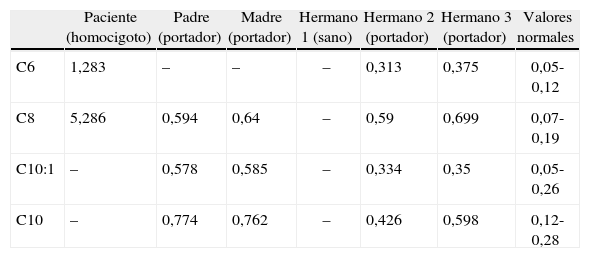
Resultado del estudio de ácidos orgánicos plasmático de la paciente y su familia
| Paciente (homocigoto) | Padre (portador) | Madre (portador) | Hermano 1 (sano) | Hermano 2 (portador) | Hermano 3 (portador) | Valores normales | |
| C6 | 1,283 | – | – | – | 0,313 | 0,375 | 0,05-0,12 |
| C8 | 5,286 | 0,594 | 0,64 | – | 0,59 | 0,699 | 0,07-0,19 |
| C10:1 | – | 0,578 | 0,585 | – | 0,334 | 0,35 | 0,05-0,26 |
| C10 | – | 0,774 | 0,762 | – | 0,426 | 0,598 | 0,12-0,28 |
Valores expresados en μmol/l.
Entre paréntesis se expone el estado (homocigoto/portador/sano) con respecto a la mutación G985A. Obsérvense los niveles muy elevados en la paciente, normales en el hermano sano y discretamente incrementados en los portadores.
C6: hexanoilcarnitina; C8: octanolicarnitina; C10:1: decenoilcarnitina; C10: decanoilcarnitina.
El déficit de MCAD afecta a 1 de cada 10.000 habitantes (1:6.500-17.000), con predominio en caucasianos2–5. Se hereda con patrón autosómico recesivo y en más del 90 % de los casos se debe a la mutación G985A3,6. La frecuencia de portadores de la misma en europeos de raza blanca oscila entre 1 de cada 40 y 1 de cada 552,4, aunque se ha observado en 1 de cada 17 individuos7 en un estudio español realizado en la etnia gitana.
Se manifiesta generalmente durante los primeros 2 años de vida, con episodios de hipoglucemia hipocetósica y encefalopatía metabólica aguda desencadenados por períodos de ayuno prolongados o enfermedades intercurrentes. En tales circunstancias se produce un aumento de las demandas energéticas, con lo que se agotan antes las reservas de glucógeno y se recurre al metabolismo de los ácidos grasos que, al estar bloqueado, conduce a hipoglucemia, acumulación de metabolitos tóxicos y disminución secundaria de las concentraciones plasmáticas de carnitina con aumento de la concentración de carnitina esterificada en orina. Dejada a su evolución natural podría tener una tasa de mortalidad del 20–25%5,8, habiéndose demostrado que un diagnóstico precoz modifica drásticamente su curso, previniendo casos del síndrome de la muerte súbita del lactante (SMSL) e importantes secuelas neurológicas5,8,9. Ya está incluida en el cribado neonatal sistemático en partes de Europa, Estados Unidos y Australia5. En España, se realiza en Galicia y, próximamente, en Murcia y Andalucía dentro del cribado neonatal expandido.
La actitud diagnóstica ante un lactante con hipoglucemias de origen incierto debe ser realizar en el momento agudo una analítica básica de orina, hemograma y bioquímica sanguínea, un estudio hormonal, aminoácidos y ácidos orgánicos en sangre y orina, y estudio de carnitina plasmática (libre y esterificada). Los hallazgos característicos de hipoglucemia hipocetósica y aciduria dicarboxílica pueden no darse en todos los casos, como en nuestra paciente, en quien se objetivaban cuerpos cetónicos en orina (presentando además valores normales de carnitina libre y esterificada en plasma), y en otros en que la aciduria dicarboxílica sólo se produce en agudizaciones. Por ello, ante un elevado índice de sospecha debe instaurarse un tratamiento y continuar el estudio diagnóstico. Una vez confirmado, el tratamiento10 consiste en evitar ayunos prolongados y triglicéridos de cadena media, administrar fécula de maíz (sobre todo antes de las horas de sueño, como forma de aporte sostenido de glucosa), carnitina (50–100mg/kg/día v.o.-v.i.) y riboflavina (100–300mg/día v.o.-v.i.).
Ante episodios de hipoglucemia de etiología incierta se debe considerar el déficit de MCAD, sobre todo si existe consaguinidad. El estudio genético y metabólico del paciente y su familia son de gran utilidad para la confirmación diagnóstica e identificación de enfermos y portadores, reduciendo la morbimortalidad asociada a esta enfermedad y permitiendo un adecuado consejo genético.
