



El síndrome de PAGOD (Pulmonary hipoplasia, hipoplasia of the pulmonary Artery, aGonadism, Omphalocele/diaphragmatic defect and Dextrocardia) es un síndrome poco frecuente1,2, que se caracteriza por la asociación de varias anomalías congénitas viscerales, incluyendo defectos cardiovasculares, hipoplasia pulmonar, defectos diafragmáticos, alteraciones genitales y la dextroposición del corazón y las estructuras mediastínicas1,3,4. Entre las alteraciones cardíacas más frecuentes, destacan la hipoplasia o atresia de la arteria pulmonar, hipoplasia de arco aórtico, hipoplasia de cavidades izquierdas, comunicación interventricular y comunicación auricular. La ambigüedad de genitales externos es frecuente. Todos los pacientes presentan hipoplasia o agenesia gonadal y alteraciones en el desarrollo de los conductos de Wolff y Muller5. Desde su descripción por Kennerknecht et al., en 1991, tan solo han sido publicados 11 casos. Su diagnóstico debe sospecharse ante la presencia de hernia diafragmática y otras malformaciones asociadas.
Presentamos el caso de un neonato de 9 días de vida, que ingresa en la Unidad de Cuidados Intensivos Pediátricos para asistencia con circulación con membrana extracorpórea veno-arterial (ECMO-VA) después de cirugía correctora de eventración diafragmática y reparación de hipoplasia de arco aórtico.
La paciente era fruto de una segunda gestación bien controlada, a término. No tenía antecedentes familiares de interés, consanguinidad ni exposición a tóxicos durante el embarazo. En la ecografía del tercer trimestre se sospechó defecto diafragmático, realizándose una resonancia magnética fetal que confirmó la existencia de eventración diafragmática (fig. 1), sin objetivarse otras anomalías. Se decidió inducción del parto, realizándose cesárea urgente por alteración del registro cardiotocográfico. Nació una niña sin esfuerzo respiratorio (Apgar 2/6) y se intubó en sala de partos.
Ante el diagnóstico de eventración diafragmática, se realizaron exploraciones complementarias para descartar malformaciones asociadas. La ecocardiografía mostró comunicación interauricular de 15mm, comunicación interventricular pequeña e hipoplasia de arco aórtico con válvula aórtica bivalva. El electrocardiograma fue compatible con síndrome de Wolf-Parkinson-White. No se objetivaron testes ni otra estructura gonadal en la ecografía abdominogenital, no se detectó ninguna anomalía en el hígado, el bazo o los riñones. Cariotipo 46XY.
La paciente presentó insuficiencia respiratoria aguda hipoxémica grave de forma progresiva. Se estabilizó y a los 9 días de vida se realizó cirugía de la eventración diafragmática (mediante toracotomía lateral) y corrección de la hipoplasia de arco (a través de esternotomía media con circulación extracorpórea). Presentó múltiples arritmias y disfunción severa del ventrículo derecho, siendo necesario soporte con ECMO-VA.
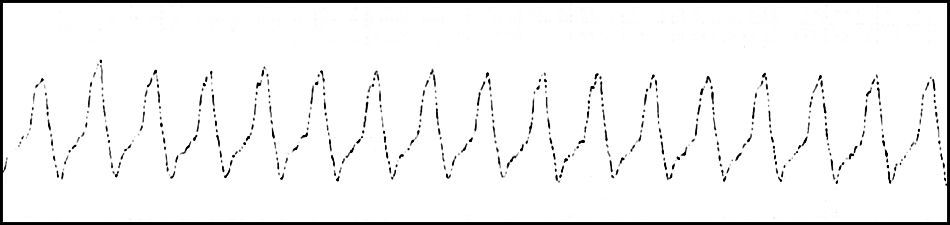
Posteriormente, presentó mala evolución a nivel respiratorio a pesar del soporte con ECMO-VA, tratamiento con óxido nítrico, epoprostenol, surfactante y ventilación de alta frecuencia oscilatoria. A nivel hemodinámico, presentó múltiples episodios de taquicardia ventricular y fibrilación ventricular (fig. 2), realizándose estudio electrofisiológico en 2 ocasiones, en los que se ablacionaron 2 vías de reentrada auriculoventricular antidrómica. La paciente persistió con arritmia intratable, disfunción biventricular severa y fallo multiorgánico progresivo. Dadas las lesiones asociadas y la mala evolución, se decidió retirar los tratamientos de soporte vital de acuerdo con la familia, se limitó el esfuerzo terapéutico. La paciente falleció a los 36 días de vida. Se realizó el estudio autópsico para documentar las malformaciones e intentar esclarecer el diagnóstico.
El estudio anatomopatológico y el análisis de anomalías asociadas (eventración diafragmática, hipoplasia pulmonar, cardiopatía congénita con anomalías de los grandes vasos y arritmias, síndrome de regresión testicular con agonadismo) fueron compatibles con síndrome de PAGOD.
La etiología del síndrome de PAGOD no está establecida, sugiriéndose un error en el desarrollo temprano de la embriogénesis. La mayor parte de los casos descritos son esporádicos, excepto los primeros casos reportados por Kennerknecht et al.1, en los que se indicaba una herencia autosómica recesiva. Algunos estudios en animales se indican el déficit de vitamina A o ácido retinoico2,6. Gavrilova et al.2 describieron un paciente con síndrome de PAGOD y niveles bajos de vitamina A en plasma al tercer día de vida, con niveles de vitamina A maternos normales posparto. En el caso de nuestra paciente, no se determinaron niveles de vitamina A en plasma.
El diagnóstico es de exclusión, siendo necesario descartar anomalías cromosómicas tipo tetrasomía 12p, trisomía 18, trisomía 22, deleción 8 p23.1, trisomía 1q, monosomía 9p, monosomía 4p y otros mediante la realización de un cariotipo7, así como otras microanomalías cromosómicas detectadas con cariotipo molecular.
El pronóstico de estos pacientes es malo, con una esperanza de vida limitada y elevada mortalidad durante la infancia4, principalmente por las anomalías cardíacas, las arritmias (hasta en un 25% asocian síndrome de Wolf-Parkinson-White4,6) y la hipoplasia pulmonar.
Es importante el conocimiento de este síndrome por parte de pediatras, neonatólogos, cardiólogos y genetistas porque, a pesar de su baja incidencia, implica una elevada morbimortalidad en el primer año de vida.
