


Sr. Editor:
La asociación VACTERL se encuadra dentro del concepto de asociación de alta frecuencia, que se entiende como un conjunto de múltiples anomalías, que no pueden ser catalogadas como un síndrome, que se presentan en al menos dos individuos y que no se deben al azar. Su herencia, por tanto, no sigue las leyes mendelianas sino que es de carácter esporádico, aunque la aparición de varios casos en una misma familia sugiere una herencia de carácter autosómico1,2.
Responde al acrónimo de defectos Vertebrales, malformaciones Anales, anomalías Cardíacas congénitas, alteraciones Traqueo-Esofágicas, malformaciones Renales y alteraciones en las extremidades ("Limbs" en inglés), fundamentalmente de la zona radial. Para su diagnóstico se requiere la presencia de, al menos, tres de los siete criterios enumerados.
Presentamos el caso de un recién nacido con un cuadro polimalformativo, constituido por las siete características mayores, así como por otras menores, algunas no descritas hasta el momento.
Se trata de un recién nacido resultado de una gestación sin controlar, durante la que se consumieron tanto tabaco como alcohol. Veinticuatro horas antes del parto se realizó la única ecografía prenatal, en la que se observó a un feto varón de 30 semanas, que eran discordantes con las 36 resultantes de la exploración física neonatal.
El parto fue vaginal, extramuros, atendido en un centro de salud cercano, y se estimó un Apgar 1/7.
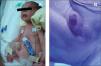
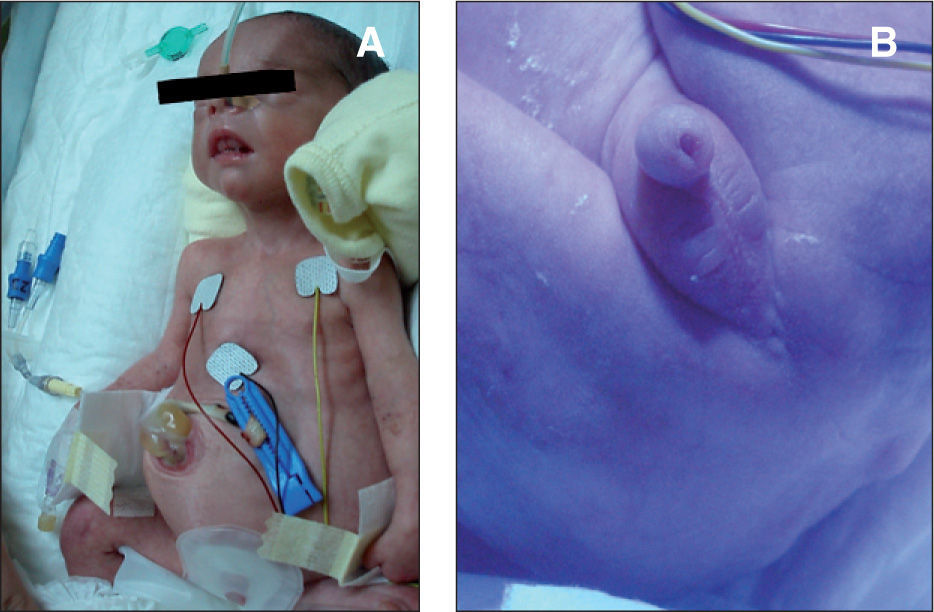
El recién nacido presentaba en el momento de su ingreso un abdomen globuloso con diástasis de rectos y hernia umbilical (fig. 1A), criptorquidia bilateral y atresia anal (fig. 1B), así como un pulgar de implantación proximal, sin ningún rasgo fenotípico facial característico.
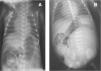
En la auscultación cardíaca se evidenció un soplo sistólico rudo III/VI en el cuarto espacio intercostal. El electrocardiograma y la radiografía torácica fueron normales. En la ecocardiografía se detectó una comunicación interventricular (CIV) perimembranosa y una comunicación interauricular (CIA) tipo ostium secundum.
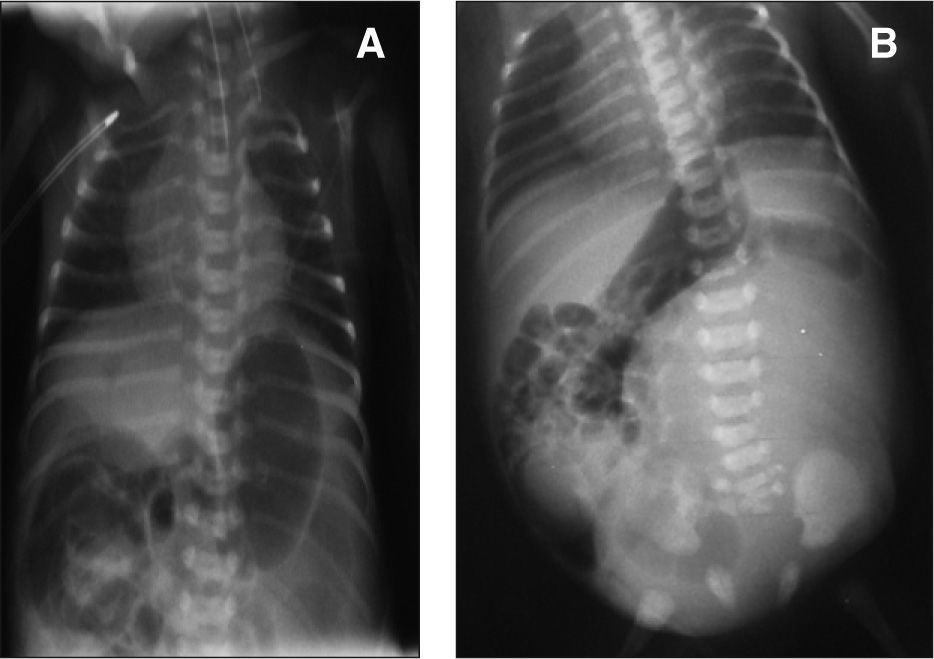
En la evaluación inicial se observó incapacidad para introducir la sonda nasogástrica a través del esófago, y que ésta se quedaba retenida en un bolsón esofágico superior, lo que concordaba con el diagnóstico de atresia esofágica; en el el estudio radiológico posterior se observó aire en el hemiabdomen derecho, y se detectó una fístula traqueoesofágica distal (fig. 2A). La ecografía abdominal detectó ureterohidronefrosis izquierda, sin visualizarse el riñón derecho.
Fue intervenido a las 48h de vida para realizar una colostomía de descarga; se evidenció un intestino delgado malrotado y parcialmente volvulado, con un ciego y un apéndice en la fosa ilíaca izquierda y, además, de la ureterohidronefrosis izquierda, por encima, había un uréter dilatado sin parénquima renal ni comunicación distal.
En el estudio radiológico vertebral se detectó agenesia del cóccix y de la quinta vértebra sacra, así como displasia de la tercera y de la cuarta vértebras de esta misma región (fig. 2B). El cariotipo fue normal.
En su evolución presentó apneas y desaturaciones frecuentes asociadas con bradicardias importantes, que al principio respondieron a las medidas básicas de reanimación, aunque falleció el quinto día por parada cardiorrespiratoria.
En el estudio autópsico, además de lo expuesto, se evidenció una comunicación ureterocloacal y la presencia de bazo supranumerario que, según nuestros conocimientos, no se había descrito hasta la fecha.
Esta entidad fue descrita, y su nombre acuñado por primera vez, por Quan-Smith en 1972, quienes sugirieron el uso del acrónimo VATER, en el que la letra R correspondía a displasias Radiales3. Fue finalmente en el año 1975 cuando se adopta el término VACTERL, que continúa en uso hasta este momento4.
Su incidencia es de 1,6 por cada 10.000 recién nacidos vivos, y tiene predilección por el sexo masculino (2,6:1).
Su patogenia consiste en una agresión mesodérmica durante la cuarta-sexta semana del desarrollo embrionario que condiciona diversas alteraciones tisulares que originan las manifestaciones clínicas descritas. Este daño se vinculó a una citopatía mitocondrial, aunque posteriormente se ha comprobado que no es una causa frecuente de la enfermedad5,6. Múltiples teratógenos pueden contribuir a esta alteración; los más reconocidos son la diabetes materna, la ingestión de hidantoína, fenitoína, estrógenos, progesterona y adriamicina durante la gestación, o la exposición elevada al plomo3,7–9. En nuestro caso, el consumo materno de alcohol podría haber contribuido, aunque no existen estudios que lo vinculen de forma irrefutable con esta asociación.
Además de los rasgos mayores, se presentan con una frecuencia variable distintos rasgos clínicos menores. Nuestro caso presentaba entre ellos criptorquidia bilateral, ureterohidronefrosis, agenesia renal derecha, fístula ureterocloacal, hernia umbilical, diastasis de rectos y bazo supranumerario, hecho este último que no hemos encontrado descrito en la bibliografía médica hasta la fecha.
Otra variante es la asociación VACTERL-H (síndrome de Briard-Evans), que añade hidrocefalia, hecho que confiere peor pronóstico y que se ha asociado a cuadros graves de anemia de Fanconi10.
En el diagnóstico diferencial de este cuadro se incluyen cromosomopatías como la trisomía 18 y las deleciones 13q-, 4q- y 6q-3.
El pronóstico es muy pobre, ya que fallece el 50–85 % de los niños en el primer año de vida y sólo sobrevive un 12 %.
Es importante tener en cuenta que la mayoría de estos niños tiene una función cerebral normal, por lo que no se deben escatimar esfuerzos tanto quirúrgicos como de rehabilitación.
