







La enfermedad oncohematológica continúa siendo la primera causa de mortalidad no traumática en la infancia y una importante causa de morbilidad. El paciente menor de 18 meses presenta particularidades clínicas, diagnósticas y terapéuticas que es interesante conocer por todo pediatra, con el fin de lograr una mayor supervivencia y una menor comorbilidad a lo largo de su vida.
Material y métodosEstudio descriptivo retrospectivo de variables clínicas, diagnósticas y terapéuticas en pacientes menores de 18 meses diagnosticados de enfermedad oncohematológica que reciben quimioterapia en una unidad de oncología pediátrica entre enero 2007 y agosto 2019.
ResultadosSetenta y dos pacientes fueron diagnosticados de 76 neoplasias que precisaron quimioterapia. La neoplasia de mayor incidencia fue la leucemia (21 pacientes), seguida del neuroblastoma (15 pacientes) y los tumores del sistema nervioso central (12 pacientes). La presentación con «síntomas amenazantes para la vida» tuvo lugar en el 20,8% de los afectados, especialmente en tumores de estirpe neural (13/15). El 18% de pacientes no presentaron síntomas al inicio. El 51% de los diagnósticos totales tuvieron lugar en «estadios avanzados». Concretamente en el caso de los tumores sólidos, el 23,6% de los inicios presentaron metástasis. Se aislaron importantes porcentajes de alteraciones genéticas implicadas en la etiopatogenia de las diferentes neoplasias.
ConclusionesEl cáncer en la primera etapa de la vida supone un reto diagnóstico y terapéutico por su diversidad fenotípica, su carga genética y sus dificultades terapéuticas. El conocimiento de sus particularidades es fundamental para un abordaje precoz y eficaz.
Oncological-haematological disease continues to be the first cause of non-traumatic mortality in childhood, as well as a significant cause of morbidity. The patient less than 18-months-old has special clinical, diagnostic, and therapeutic features that all paediatricians are interested in determining, with the aim of achieving greater survival and a lower morbidity throughout the lives of their patients.
Material and methodsA retrospective, descriptive study was carried out using the clinical, diagnostic, and therapeutic variables in patients less than 18-months-old diagnosed with an oncological-haematological that received chemotherapy in a Paediatric Oncology Unit between January 2007 and August 2019.
ResultsA total of 72 patients were diagnosed with 76 cancers that required chemotherapy. The most common cancer was leukaemia (21 patients), followed by neuroblastoma (15 patients), and tumours of the central nervous system (12 patients). The presentation of “life-threatening symptoms” was seen in 20.8% of cases, particularly in tumours of neural origin (13/15). Although 18% of patients showed no symptoms on diagnosis, just over half (51%) of the diagnoses took place in the “advanced stages”. Particularly in the case of solid tumours in which 23.6% were diagnosed with metastases. A significant percentage of genetic alterations implicated in the aetiopathogenesis of the different cancers were found.
ConclusionsCancer in the first stages of life is a diagnostic and therapeutic challenge due to its phenotypical diversity, its genetic load, and its therapeutic difficulties. Knowledge of its particular features is essential for its early and effective approach.
El cáncer pediátrico es una enfermedad de amplia trascendencia médica, psicológica y social.
Es la primera causa de mortalidad no traumática en la infancia con una incidencia anual de 155,5 nuevos casos por millón, lo que supone 1.100 nuevos casos de cáncer infantil al año en España1,2.
La propia enfermedad oncológica y los tratamientos antineoplásicos pueden provocar morbilidad a lo largo de toda la vida del paciente, especialmente cuando el cáncer aparece en las edades más tempranas. Un diagnóstico precoz permite disminuir la agresividad de los tratamientos y aumentar las probabilidades de curación, por lo que el conocimiento de las características clínicas de esta enfermedad por parte del pediatra es fundamental3,4.
El diagnóstico y el tratamiento del cáncer durante los primeros meses de vida puede ser especialmente complicado ya que, dada la inmadurez del organismo y su constante desarrollo, presenta peculiaridades que lo diferencian de otras etapas infantiles.
En primer lugar, la naturaleza tumoral
Según los datos proporcionados por el Registro Español de Tumores Infantiles (RETI)5, el cáncer infantil más frecuente es la leucemia aguda (30%), en segundo lugar, los tumores del sistema nervioso central (TSNC) (21,7%) y seguidamente los linfomas (12,7%). Sin embargo, en época lactante y durante la primera infancia los tumores embrionarios cobran especial relevancia, siendo raros los carcinomas. De hecho, en algunas series la prevalencia de la leucemia es superada por la del neuroblastoma y muy cercana a la de los TSNC embrionarios como los astrocitomas y los meduloblastomas. Otros tumores embrionarios de alta incidencia relativa son el retinoblastoma, el hepatoblastoma y el nefroblastoma o tumor de Wilms (TW)1.
En segundo lugar, la expresión sistémica de la neoplasia
Tanto por las características fisiológicas del lactante, como por la dificultad en la comunicación de la sintomatología inicial, es relativamente frecuente que las manifestaciones clínicas al diagnóstico sean multifactoriales y/o de características agresivas, consecuencia de la alta carga tumoral.
En tercer lugar, los factores genéticos
Se estima que el 5-10% del cáncer es hereditario6. Suelen aparecer de forma precoz respecto a sus homólogos no hereditarios, frecuentemente lo hacen en los primeros años de vida y son de comportamiento más agresivo. Tal es el caso de algunos pacientes con retinoblastoma (gen RB) o TW (gen WT1).
Otro factor genético típicamente implicado en la carcinogénesis del paciente lactante es la positividad del gen MLL en leucemias, el cual confiere mal pronóstico a la enfermedad.
Por último, las limitaciones terapéuticas
El riesgo quirúrgico y anestésico de los lactantes es mayor. Así mismo, la quimioterapia puede provocar mayor toxicidad a lo esperado, dadas las particularidades farmacocinéticas en este grupo de edad. Es conocido que el potencial de desarrollo neurológico y hormonal puede verse negativamente afectado por tratamientos como la radioterapia, por lo que su empleo en menores de 3 años es excepcional.
En esta revisión se estudiarán las características clínicas, diagnósticas y terapéuticas de pacientes oncológicos receptores de quimioterapia y diagnosticados de cáncer en sus 18 primeros meses de vida, analizando las características diferenciales con respecto al resto de grupos pediátricos.
Pacientes y métodosEstudio descriptivo retrospectivo de los pacientes menores de 18 meses diagnosticados de enfermedad oncológica que reciben tratamiento quimioterápico en el Hospital La Fe de Valencia entre enero 2007 y agosto 2019.
Los tumores analizados fueron leucemias agudas, TSNC, neuroblastomas, nefroblastomas, hepatoblastomas, retinoblastomas y sarcomas.
Se estudiaron las siguientes variables, tanto de manera conjunta en la población del estudio como dividida según cada tipo de neoplasia:
- a)
Sexo del paciente. Cualitativa
- b)
Edad al diagnóstico. Cuantitativa
- c)
Clínica de presentación. CualitativaSe enumeran los síntomas clasificados por el aparato o sistema al que afectan, así como su agresividad. Con el fin de unificar los síntomas de los diferentes tumores se ha decidido categorizar la clínica al inicio como «síntomas amenazantes para la vida» (SAV)7 en caso de cumplimiento de los criterios pertinentes. Dicha categorización, expuesta en la tabla 1, es aplicada típicamente al neuroblastoma y se encuentra recogida en los protocolos International Society of Paediatric Oncology (SIOP).
Tabla 1.Síntomas amenazantes para la vida
1. Tumor intraespinal con síntomas compresivos, ocupación de más de 1/3 del canal raquídeo, señal anormal oborramiento de espacios leptomeníngeos 2. Afectación del estado general: Dolor que requiere opiáceos Gastrointestinal Vómitos que requieren vía intravenosa o sonda nasogástrica Pérdida de >10% de peso Diarrea por VIP que no responde a quimioterapia Respiratorio Distrés de causa no infecciosa Taquipnea >60 Necesidad de oxígeno Asistencia respiratoria Cardiovascular Hipertensión Compresión de vena cava inferior ± edemas de miembros inferiores Renal Disfunción renal, creatinina X2 respecto al límite superior de la normalidad Hidrouréter/hidronefrosis Hepático Disfunción hepática >2 respecto al límite superior de la normalidad Coagulación intravascular diseminada Trombocitopenia <50.000/mcl Disfunción vesical o intestinal secundaria a efecto masa 3. Un volumen tumoral suficientemente grande que supone riesgo de rotura o afectación sistémica aguda VIP: péptido intestinal vasoactivo.
- d)
Localización del tumor primario. Cualitativa
- e)
Extensión o metástasis. Cualitativa
- f)
Histología o morfología celular. Cualitativa
- g)
Alteraciones genéticas. Cualitativa
- h)
Estadio de la enfermedad según diferentes protocolos en función del tipo de tumor (tabla 2). Cuantitativa
Tabla 2.Protocolos utilizados para clasificación de neoplasias y estadios considerados como «avanzados»
Neoplasia Protocolo/Staging system Estadio «avanzado» Leucemia a) 0-12 meses Protocolo LAL-LACTANTES SHOP-2002 Alto riesgo o muy alto riesgo b) 12-18 meses Protocolo LAL/SEHOP PETHEMA 2013 Alto riesgo Neuroblastoma International Neuroblastoma Risk Group Staging System Metastásico M+ (excluye MS) Retinoblastoma Clasificación Reese-Ellsworth Estadio III, IV o V Tumores SNC Localizado o diseminado Diseminado Tumor de Wilms Protocolo SIOP UMBRELLA Estadio IV o V Hepatoblastoma Clasificación PRETEXT Estadio IV o metastásico Sarcoma Rabdomiosarcoma no mts EpSSG RM-2005 no metastásico Alto riesgo o muy alto riesgo Rabdomiosarcoma metastásico Protocolo de IRSG Metastásico Sarcoma de Ewing Localizado o metastásico EpSSG: European paediatric Soft tissue sarcoma Study Group; IRSG: Intergroup Rhabdomyosarcoma Group; LAL: Leucemia linfoblástica aguda; PETHEMA: Programa Español de Tratamientos de Hematología; RM: rabdomiosarcoma; SHOP: Sociedad Oncohematología Pediátrica; SIOP: International Society Paediatric Oncology; SNC: sistema nervioso central.
- i)
Se unifica la estadificación clasificando los tumores en «estadio avanzado» o «estadio no avanzado» bajo los criterios indicados en la tabla 2
Las variables cualitativas se describieron con el recuento numérico (porcentaje) de cada una de ellas. Las variables cuantitativas como mediana ± rango intercuartílico ante ausencia de normalidad en la distribución de los datos (p<0,05, prueba de Kolmogorov-Smirnov).
El análisis estadístico se realizó con el programa Excel® y las gráficas con Excel®, Microsoft Office Publisher® y Power Point®.
ResultadosVariables generalesEn el periodo analizado (enero 2007-agosto 2019) fueron diagnosticados 72 pacientes de enfermedad oncológica que precisaron quimioterapia en el Hospital La Fe. Cuatro de ellos presentaron más de una neoplasia, sumando un total de 76 en dicho periodo.
La neoplasia de mayor incidencia fue la leucemia (21 pacientes), seguida del neuroblastoma (15 pacientes), los TSNC central (12 pacientes), el retinoblastoma (9 pacientes, 13 neoplasias), TW (7 pacientes), el hepatoblastoma (5 pacientes) y el sarcoma (3 pacientes). En conjunto, el 70,8% de los pacientes fue diagnosticado de tumor sólido (51/72) frente al 29,2% diagnosticados de leucemia (21/72).
La mediana de edad al diagnóstico fue de 9,5 meses, con ligero predominio del sexo masculino (54,2%) frente al femenino (45,8%).
Variables clínicasEn referencia a la presentación clínica de la enfermedad oncológica, es variable según el tipo de enfermedad, tal y como muestran la figura 1.
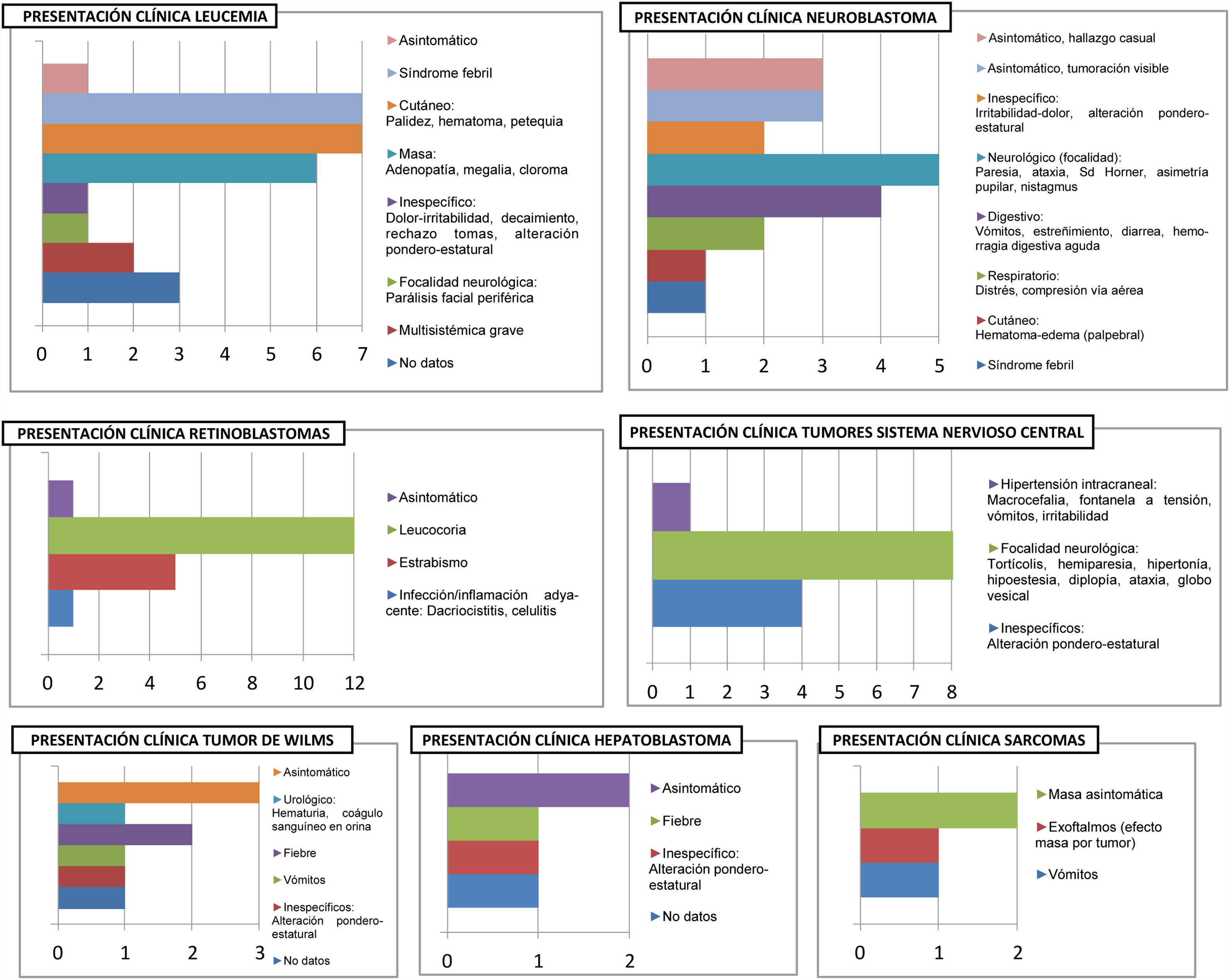
Trece pacientes (18,1% del total) se encontraban asintomáticos al diagnóstico: En 8 casos fue un hallazgo incidental y en 5 el motivo que llevó a la consulta médica fue una masa asintomática: Tal es el caso de 3 pacientes afectos de neuroblastomas (forma de presentación del 20% de estos tumores) y 2 diagnosticados de sarcoma. Por el contrario, todos los pacientes diagnosticados de TSNC (los 12) presentaron síntomas al inicio, principalmente síntomas focales.
Analizando la gravedad, cabe destacar que 15 pacientes iniciaron con SAV (20,8%), siendo la mayoría de los tumores de origen neural: 7 casos de tumores SNC (46,6% de los SAV) y 6 casos de tumores del SNP tipo neuroblastoma (40% de los SAV). El resto de los SAV se presentaron en 2 pacientes afectos de leucemia, los cuales presentaron distrés respiratorio y oliguria secundarios al síndrome de lisis tumoral, precisando ingreso en unidad de cuidados intensivos pediátricos (UCIP).
En las siguientes tablas se recogen la distribución de tumores que inician con SAV (tabla 3) y se concreta la sintomatología (tabla 4).
Tumores que comienzan con SAV y su sistema implicado
| Neoplasia | Presentación clínica que supone «SAV» | Sistema implicado |
|---|---|---|
| Neuroblastoma | Paresia miembros inferiores | Neurológico |
| Neuroblastoma | Distrés respiratorio | Respiratorio |
| Neuroblastoma | Hemorragia aguda de origen abdominal | Digestivo |
| Neuroblastoma | Vómitos + inestabilidad + ataxia + nistagmo | Neurológico |
| Neuroblastoma | Paresia miembros inferiores + síndrome de Horner | Neurológico |
| Neuroblastoma | Masa cervical + compresión vía aérea | Respiratorio |
| LLA | Taquipnea + oliguria + petequias + esplenomegalia | Respiratorio + renal |
| LMA | Distrés respiratorio + oliguria + disminución conciencia | Respiratorio + renal + neurológico |
| Tumor SNC | Tortícolis + hipotonía ESD | Neurológico |
| Tumor SNC | Irritabilidad + globo vesical + estreñimiento | Neurológico |
| Tumor SNC | Distensión abdominal + dolor + anorexia | Neurológico |
| Tumor SNC | Alteración marcha + desequilibrio | Neurológico |
| Tumor SNC | Paresia miembros inferiores | Neurológico |
| Tumor SNC | Paresia miembros inferiores | Neurológico |
| Tumor SNC | Alteración marcha + hipertonía + vómitos | Digestivo + urológico |
ESD: extremidad superior derecha; LLA: leucemia linfoblástica aguda; LMA: leucemia mieloblástica aguda; SAV: síntomas amenazantes para la vida; SNC: sistema nervioso central.
El 45,8% de los pacientes (33/72) presentaron un estadio tumoral «avanzado» desde el diagnóstico (tabla 2).
Analizando los tumores sólidos, 17 presentaron metástasis al inicio (23,6% del total), 12 casos (16,6%) si excluimos los 5 casos con neuroblastoma MS.
A continuación se expone la extensión tumoral, el diagnóstico histológico y la estadificación de los 3 grupos tumorales sólidos más frecuentes de la muestra de estudio: El retinoblastoma, el neuroblastoma y los TSNC (tabla 5).
Extensión tumoral, diagnóstico histológico y estadificación del retinoblastoma, neuroblastoma y tumores del SNC
| Neoplasia | Localización primaria | Histología | Metástasis | Estadificación |
|---|---|---|---|---|
| Rb | Ojo derecho | Neuroectodérmico | No | III |
| Rb | Ojo derecho | Neuroectodérmico | No | V |
| Rb | Ojo derecho | Neuroectodérmico | No | III |
| Rb | Ojo izquierdo (ambos) | Neuroectodérmico | No | III |
| Rb | Ojo derecho (ambos) | Neuroectodérmico | No | V |
| Rb | Ojo izquierdo | Neuroectodérmico | No | III |
| Rb | Ojo izquierdo (ambos) | Neuroectodérmico | No | IV |
| Rb | Ojo derecho (ambos) | Neuroectodérmico | No | IV |
| Rb | Ojo izquierdo | Neuroectodérmico | No | II |
| Rb | Ojo izquierdo | Neuroectodérmico | No | IV |
| Rb | Ojo izquierdo (ambos) | Neuroectodérmico | No | III |
| Rb | Ojo derecho (ambos) | Neuroectodérmico | No | V |
| Rb | Ojo izquierdo | Neuroectodérmico | No | II |
| Nb | Abdominal | Diferenciado | No | L2 |
| Nb | Abdominal | Indiferenciado | Hígado | MS |
| Nb | Abdominal | Indiferenciado | No | L2 |
| Nb | Torácico | Pobremente diferenciado | Hígado | MS |
| Nb | Cérvico-torácico | Pobremente diferenciado | No | L1 |
| Nb | Abdominal | Pobremente diferenciado | Hígado | MS |
| Nb | Abdominal | Diferenciado | Hígado + subpleural | M |
| Nb | Abdominal | Pobremente diferenciado | No | L1 |
| Nb | Abdominal | Diferenciado | Piel | MS |
| Nb | Abdominal | Indiferenciado | No | L2 |
| Nb | Tóraco-abdominal | Pobremente diferenciado | SNC | L2 |
| Nb | Abdominal | Diferenciado | No | L1 |
| Nb | Abdominal | Indiferenciado | Hueso | M |
| Nb | Abdominal | Diferenciado | Hïgado | MS |
| Nb | Cervical | Indiferenciado | No | L2 |
| SNC | Hipotálamo | Astrocitoma pilocítico | No | Localizado |
| SNC | Bulbo-medular | PNET | No | Localizado |
| SNC | Masa presacra | Germinativo mixto | No | Localizado |
| SNC | Masa pélvica | Germinativo: teratoma inmaduro | No | Localizado |
| SNC | Óptico-hipotalámico | Astrocitoma pilocítico | Pioaracnoidea + espinal | Diseminado |
| SNC | Fosa posterior | Meduloblastoma nodular | No | Localizado |
| SNC | Fosa posterior | PNET | No | Localizado |
| SNC | Fosa posterior | Meduloblastoma nodular | Duramadre | Diseminado |
| SNC | Médula sacro | Germinativo: teratoma inmaduro | Medular presacra | Diseminado |
| SNC | Médula | Astrocitoma pilocítico | Medular dorso-lumbar | Diseminado |
| SNC | Fosa posterior | Meduloblastoma nodular | Ventrículos cerebrales | Diseminado |
| SNC | Médula | Rabdoide | No | Localizado |
Nb: neuroblastoma; PNET: primitive neuroectodermal tumor; Rb: retinoblastoma; SNC: sistema nervioso central.
En referencia a algunos marcadores genéticos de riesgo en enfermedad tumoral pediátrica, los resultados son los siguientes:
- 1.
LLA B y fusiones genéticas que involucren al gen MLL (11q23): Presente en el 46% de los casos (6/13), todos implican leucemias de muy alto riesgo.
- 2.
Neuroblastoma y amplificación nMYC8–10: Presente en el 6,6% de los casos (1/15). El paciente afecto comenzó con hemorragia retroperitoneal consecuencia de rotura tumoral que condicionó ingreso en la UCIP. Recibió tratamiento quirúrgico y quimioterápico según protocolo HR-NEUROBLASTOMA 1.8/SIOPEN, pese a lo cual sufrió una recaída a los 15 meses del diagnóstico y 5 meses después.
- 3.
Las alteraciones cromosómicas11,12 fueron registradas en 8 pacientes (53,3% de los neuroblastomas): 3 alteraciones cromosómicas segmentarias (ACS) (37,5%) y 5 alteraciones cromosómicas numéricas (ACN) (62,5%).
- 4.
Retinoblastoma y mutación gen RB113: En la presente serie se describen 10 pacientes con retinoblastoma que presentan un total de 13 tumores. Esto es, 3 pacientes presentan retinoblastoma bilateral (3/10, 30%), enviados a consejo genético. En uno de ellos se registra la mutación (c224G>A) en el exón 2 del gen RB1.
El tratamiento quimioterápico ha sido el criterio de inclusión. Ahora bien, no ha sido el único tratamiento recibido por los pacientes, tal y como se muestra en la figura 2.
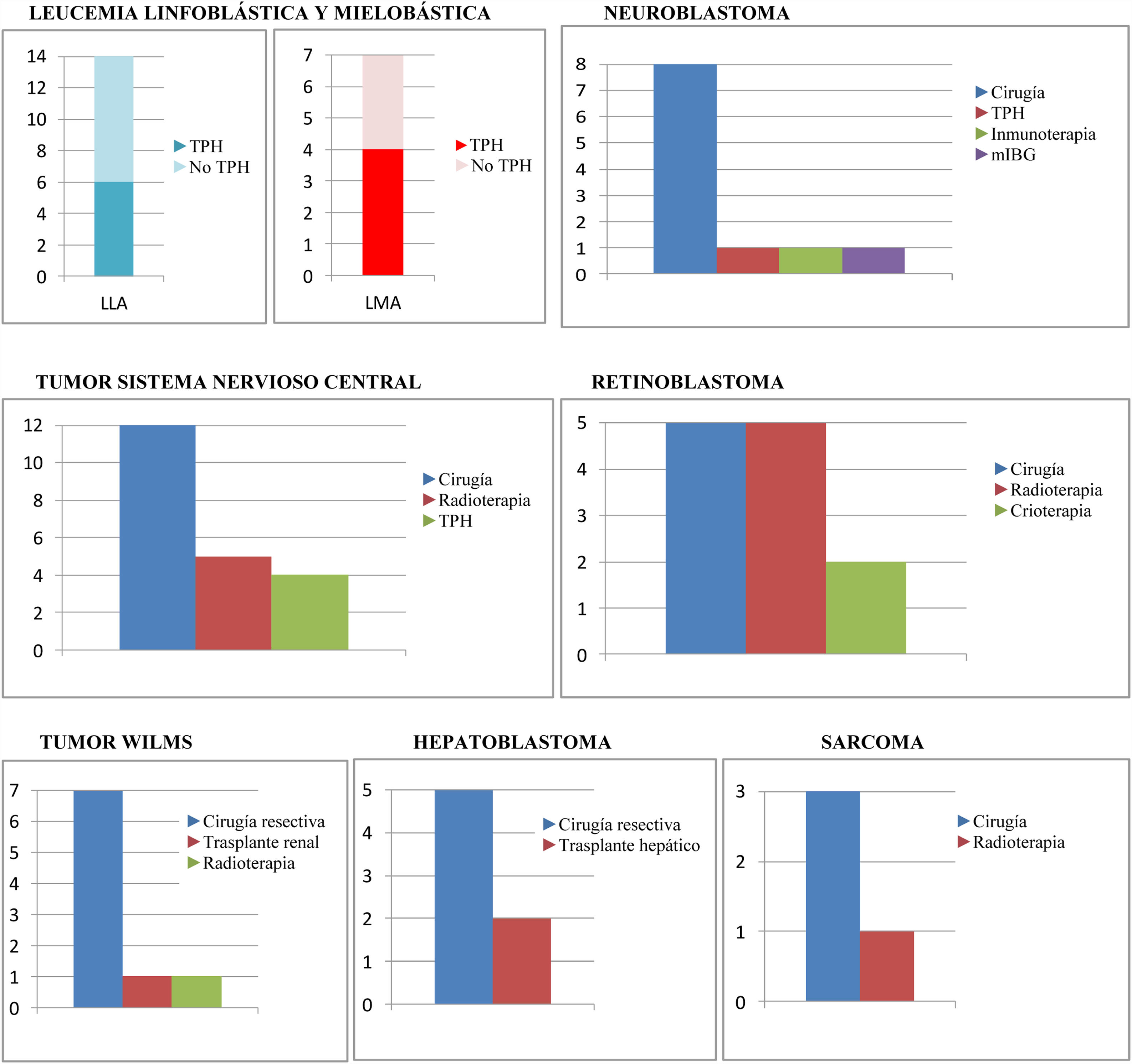
Tratamientos concomitantes al tratamiento quimioterápico en los diferentes grupos tumorales.
LLA: leucemia linfoblástica aguda; LMA: leucemia mieloblástica aguda; mIBG: gammagrafía con radioisótopo de metayodo; No TPH: no trasplante de progenitores hematopoyéticos; TPH: trasplante de progenitores hematopoyéticos.
El diagnóstico y tratamiento del cáncer en la primera etapa de la vida resulta un reto para los profesionales que trabajamos con pacientes pediátricos. Su precocidad tiene importantes implicaciones en la supervivencia y en la comorbilidad de estos pacientes, por lo que conocer las particularidades de este grupo poblacional resulta clave para su adecuado manejo.
Este estudio analiza la experiencia de nuestro centro en la presentación clínica, diagnóstico y tratamiento de los pacientes menores de 18 meses de edad con enfermedad oncológica que hayan recibido tratamiento quimioterápico. Para ello se han revisado retrospectivamente los casos de nuestra unidad durante más de 12 años (desde enero 2007 hasta agosto 2019).
Características generalesLa distribución tumoral es similar a la descrita por la literatura, siendo la leucemia la neoplasia más frecuente. Cabe destacar que la mediana de edad de los pacientes diagnosticados de leucemia es 11 meses (RI 9), superior a la de los tumores embrionarios (neuroblastoma, TSNC embrionarios, TW, hepatoblastoma y retinoblastoma), cuya mediana de edad es de 8,5 meses (RI 8), apoyando el hecho de que los tumores embrionarios son especialmente prevalentes en los primeros meses de vida.
La distribución por sexo también concuerda con la literatura, con un leve predominio del sexo masculino (54,2%) frente al femenino (45,8%).
Los resultados evidencian la amplia variedad de sintomatología de estos pacientes (fig. 1), así como los datos de afectación multifactorial (tabla 4) y su frecuente presentación agresiva:
- a)
En nuestra muestra, el 20,8% de los pacientes cumplen criterios SAV, superior a otras edades pediátricas. El grupo tumoral de presentación más agresiva lo forman los TSNC por los siguientes motivos: En primer lugar, supone el 46,6% de los casos con SAV (86,6% si consideramos todas las neoplasias del sistema nervioso, esto es, TSNC y neuroblastomas). En segundo lugar, es el único grupo tumoral que no presentó casos de pacientes asintomáticos. En tercer lugar, la clínica más frecuente fue la focalidad neurológica en forma de hemiparesia, hipertonía, diplopía, globo vesical… Otra focalidad que merece gran atención por su aparente «inocencia» es la tortícolis, forma de presentación de algunos tumores cerebrales en lactantes.
- b)
Por el contrario, en un amplio grupo de pacientes el diagnóstico tiene lugar por aparición de una masa asintomática (5 casos) o como hallazgo accidental (8 pacientes).
A continuación se comentarán algunos hallazgos de interés según grupos tumorales.
Leucemia14Según la literatura, si bien la leucemia linfoide sigue siendo la predominante en las primeras etapas de la vida, es típico que la proporción cambie a favor de estirpe mieloide y mixta15. Efectivamente, en nuestra muestra se recogen 14 pacientes con leucemia de estirpe linfoide (14/21 frente al 80% en otras edades), 6 de estirpe mieloide (6/21 frente al 20% habitualmente) y una leucemia mixta (1/21). Si consideramos únicamente nuestra población lactante (muestra <12 meses), los resultados son todavía más dispares con la población pediátrica general: 7/14 LLA, 7/14 LMA y 1/14 leucemia mixta.
El porcentaje de fusiones genéticas que involucran al gen MLL (11q23) suponen el 46% de la muestra, consecuente con la literatura16, ampliamente superior a otros grupos poblaciones.
Tumores del sistema nervioso centralLos TSNC son muy frecuentes en edad pediátrica. Habitualmente infratentoriales, en población lactante destaca su predominio supratentorial17. Probablemente este hecho justifique la alta proporción de sintomatología focal, en contra de lo observado a otras edades pediátricas (alta prevalencia de clínica secundaria a hidrocefalia). La histología más habitual es embrionaria. La corta edad es un factor de mal pronóstico en este tipo de neoplasias18.
En nuestra muestra los TSNC son los cuartos en frecuencia y con un claro predominio embrionario (66,6%).
La localización más frecuente es la médula espinal (6 casos), seguida de la fosa posterior (4 casos) y la región supratentorial (2 casos). Probablemente esta distribución atípica se deba a que los pacientes incluidos en el estudio son pacientes que reciben quimioterapia, luego excluye tumores quimiorresistentes y aquellos en los cuales la monoterapia quirúrgica es curativa. Los pacientes que recibieron radioterapia no lo hicieron hasta la edad mínima de 3 años.
Como se ha comentado en el apartado previo, los TSNC son los tumores de presentación clínica más agresiva de la muestra.
NeuroblastomaSupone la segunda neoplasia más frecuente (15 casos), cercana a las leucemias (21 casos). Es importante tener en cuenta que existen neuroblastomas cuya actitud terapéutica es únicamente quirúrgica o incluso expectante, por lo que nuestra muestra, que recoge pacientes que reciben quimioterapia, probablemente infraestime su incidencia real.
La localización más típica del neuroblastoma es abdominal, con aumento relativo de la localización torácica y cervical en el paciente lactante. En nuestra muestra el 73,3% de los tumores son abdominales, con 3 casos torácicos y 2 cervicales.
De presentación clínica muy dispar: Por un lado se recoge el 20% de los casos con diagnóstico casual o sobre una masa asintomática, y por el contrario no son infrecuentes manifestaciones de enfermedad diseminada. En el 26,6% de los casos el paciente presentó signos focales al inicio (paresia de miembros inferiores, síndrome de Horner o asimetría pupilar) secundarios a infiltración nerviosa, en el 6,6% el signo de alarma fue una tumoración subcutánea inguinal secundaria a infiltración cutánea.
Respecto al estadio tumoral, en nuestra muestra la distribución es la siguiente: L1 13,3%, L2 33,3%, M+ 20% y MS 33,3%.
El estadio MS es característico de esta población menor de 18 meses. En ausencia de parámetros genéticos que impliquen gravedad, el pronóstico de estos tumores es muy bueno pese a la infiltración a nivel hepático, esplénico, piel o médula ósea, incluso sin tratamiento19. En nuestra muestra se recogen 5 casos de neuroblastoma MS (33,3%). Los motivos que han justificado rechazar la actitud expectante han sido la presencia de ACS en 3 pacientes y la presencia de SAV en uno. El paciente restante fue diagnosticado y tratado en 2008, fecha en la cual no existía una evidencia tan firme de que la actitud expectante fuera una alternativa válida.
RetinoblastomaLos pacientes afectos de retinoblastoma constituyen los cuartos en frecuencia.
El 92% de los pacientes (12/13) presentaron leucocoria al inicio, un signo clínico de alta sensibilidad y especificidad en el diagnóstico del retinoblastoma. Enfatizar en este punto la importancia de la búsqueda del reflejo pupilar en las revisiones pediátricas ordinarias.
La aparición del retinoblastoma en lactantes debe alertarnos de un origen germinal-hereditario de mayor agresividad y extensión. En tal caso resulta importante derivar al paciente a consejo genético. En nuestra muestra destaca el alto porcentaje de bilateralidad (46,1%), así como la necesidad de tratamientos agresivos (el 38,5% de los pacientes sufren enucleación).
Tumor de WilmsEl pronóstico del TW en lactantes es mejor que en otras etapas de la vida, dado que frecuentemente su histología es epitelial y su estadio precoz20.
En nuestro estudio los 7 TW son epiteliales (100%), 3 de los cuales presentaron estadio avanzado al diagnóstico.
HepatoblastomaLo mismo podemos afirmar acerca del hepatoblastoma de la primera infancia: Suele presentar buen pronóstico consecuencia de una histología favorable.
En nuestra muestra la mayoría de los hepatoblastomas son epiteliales. Hubo un caso de histología mixta, el único paciente que comenzó con metástasis pulmonares.
Señalar que ha sido frecuente el diagnóstico del tumor como hallazgo accidental (40-50%).
SarcomasAunque muy raro a esta edad, el sarcoma más frecuente en la primera infancia es el rabdomiosarcoma. En nuestra muestra contamos con un rabdomiosarcoma orbitario y un rabdomiosarcoma en extremidad inferior (gastronemio derecho). La histología suele ser favorable, en el presente estudio ambos son embrionarios.
En conclusión, las neoplasias que afectan a los pacientes menores de 18 meses presentan diferencias importantes respecto a otros grupos pediátricos a nivel clínico, diagnóstico y terapéutico21. Tener en cuenta estas particularidades permite un diagnóstico precoz, un tratamiento más adecuado y un pronóstico más favorable, por lo que resulta fundamental su conocimiento por parte de los profesionales que trabajamos con pacientes pediátricos.
Así mismo, dada la especial vulnerabilidad de este grupo de pacientes a los tratamientos antineoplásicos, resulta muy interesante la utilización de nuevas herramientas de medicina personalizada como la farmacogenética y la farmacogenómica22–24, las cuales permiten optimizar el efecto terapéutico maximizando su eficacia y minimizando sus riesgos25.
FinanciaciónTrabajo parcialmente financiado por la Fundación Mutua Madrileña mediante “Ayudas Investigación 2016”, la Asociación Pablo Ugarte y la Asociación Esperanza y Sonrisas.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
