




Oncological-haematological disease continues to be the first cause of non-traumatic mortality in childhood, as well as a significant cause of morbidity. The patient less than 18-months-old has special clinical, diagnostic, and therapeutic features that all paediatricians are interested in determining, with the aim of achieving greater survival and a lower morbidity throughout the lives of their patients.
Material and methodsA retrospective, descriptive study was carried out using the clinical, diagnostic, and therapeutic variables in patients less than 18-months-old diagnosed with an oncological-haematological that received chemotherapy in a Paediatric Oncology Unit between January 2007 and August 2019.
ResultsA total of 72 patients were diagnosed with 76 cancers that required chemotherapy. The most common cancer was leukaemia (21 patients), followed by neuroblastoma (15 patients), and tumours of the central nervous system (12 patients). The presentation of “life-threatening symptoms” was seen in 20.8% of cases, particularly in tumours of neural origin (13/15). Although 18% of patients showed no symptoms on diagnosis, just over half (51%) of the diagnoses took place in the “advanced stages”. Particularly in the case of solid tumours in which 23.6% were diagnosed with metastases. A significant percentage of genetic alterations implicated in the aetiopathogenesis of the different cancers were found.
ConclusionsCancer in the first stages of life is a diagnostic and therapeutic challenge due to its phenotypical diversity, its genetic load, and its therapeutic difficulties. Knowledge of its particular features is essential for its early and effective approach.
La enfermedad oncohematológica continúa siendo la primera causa de mortalidad no traumática en la infancia y una importante causa de morbilidad. El paciente menor de 18 meses presenta particularidades clínicas, diagnósticas y terapéuticas que es interesante conocer por todo pediatra, con el fin de lograr una mayor supervivencia y una menor comorbilidad a lo largo de su vida.
Material y métodosEstudio descriptivo retrospectivo de variables clínicas, diagnósticas y terapéuticas en pacientes menores de 18 meses diagnosticados de enfermedad oncohematológica que reciben quimioterapia en una Unidad de Oncología Pediátrica entre enero 2007 y agosto 2019.
Resultados72 pacientes fueron diagnosticados de 76 neoplasias que precisaron quimioterapia. La neoplasia de mayor incidencia fue la leucemia (21 pacientes), seguida del neuroblastoma (15 pacientes) y los tumores sistema nervioso central (12 pacientes). La presentación con “síntomas amenazantes para la vida” tuvo lugar en el 20,8% de los afectados, especialmente en tumores de estirpe neural (13/15). 18% de pacientes no presentaron síntomas al debut. El 51% de los diagnósticos totales tuvieron lugar en “estadios avanzados”. Concretamente en el caso de los tumores sólidos, el 23,6% de los debuts presentaron metástasis. Se aislaron importantes porcentajes de alteraciones genéticas implicadas en la etiopatogenia de las diferentes neoplasias.
ConclusionesEl cáncer en la primera etapa de la vida supone un reto diagnóstico y terapéutico por su diversidad fenotípica, su carga genética y sus dificultades terapéuticas. El conocimiento de sus particularidades es fundamental para un abordaje precoz y eficaz.
Paediatric cancer is a health problem with a significant clinical, psychological and social impact.
It is the leading non-traumatic cause of death in children, with an annual incidence of 155.5 cases per million, which roughly corresponds to 1100 new cases of childhood cancer per year in Spain.1,2
Cancer itself and cancer treatments can cause morbidity throughout the life of the patient, especially in cases with onset at an early age. Early diagnosis allows the use of less aggressive treatments and increases the probability of a cure, so it is essential that paediatricians be acquainted with the clinical features of cancer in this age group.3,4
The diagnosis and treatment of cancer in the early months of life may be particularly challenging due to the immaturity and constant growth of the organism, which give rise to peculiarities that differentiate this age from other stages of childhood.
First of all, the types of tumoursBased on data from the Registro Español de Tumores Infantiles (Spanish Register of Paediatric Tumours, RETI),5 the most frequent form of cancer in the paediatric age group is acute leukaemia (30%), followed by central nervous system (CNS) tumours (21.7%) and lymphomas (12.7%). However, in infancy and early childhood embryonal tumours are particularly important, while carcinomas are rare. In fact, in some case series the incidence of leukaemia was lower compared to the incidence of neuroblastoma and similar to the incidence of CNS embryonal tumours, such as astrocytoma or medulloblastoma. Other relatively frequent embryonal tumours are retinoblastoma, hepatoblastoma and nephroblastoma or Wilms tumour (WT).1
Second, the systemic manifestations of oncologic diseaseDue to the physiological characteristics of infants and the barriers in communicating the initial symptoms, the presenting clinical features at diagnosis are often multifactorial and/or characteristic of aggressive disease due to a high tumour load.
Third, genetic factorsIt is estimated that 5% to 10% of cancer cases are hereditary.6 These cases tend to have an early onset compared to the non-hereditary cases of the same type of cancer, frequently in the early years of life and with a more aggressive course. This is the case of some patients with retinoblastoma (RB1 gene) or WT (WT1 gene).
Another genetic factor that is frequently involved in carcinogenesis in infants is the presence of KMT2A rearrangements in leukaemia cases, which is associated with a poor prognosis.
Lastly, the limitations of treatmentThe risks of surgery and anaesthesia are greater in infants. Chemotherapy may also be more toxic than expected due to the pharmacokinetic particularities in this age group. It is known that neurodevelopment and hormone production may be negatively affected by treatments such as irradiation, so their use before age 3 years is exceptional.
In the review presented here, we analysed clinical, diagnostic and treatment variables in oncological patients given a cancer diagnosis before 18 months of age and managed with chemotherapy, analysing the differences relative to other paediatric populations.
Patients and methodsWe conducted a retrospective descriptive study in patients given a diagnosis of cancer before age 18 months treated with chemotherapy at the Hospital La Fe de Valencia between January 2007 and August 2019.
The types of tumour included in the analysis were acute leukaemia, CNS tumours, neuroblastoma, nephroblastoma, hepatoblastoma, retinoblastoma and sarcoma.
We analysed the following variables in the entire sample and by type of cancer:
- a)
Sex of the patient (qualitative).
- b)
Age at diagnosis (quantitative).
- c)
Presenting symptoms (qualitative).
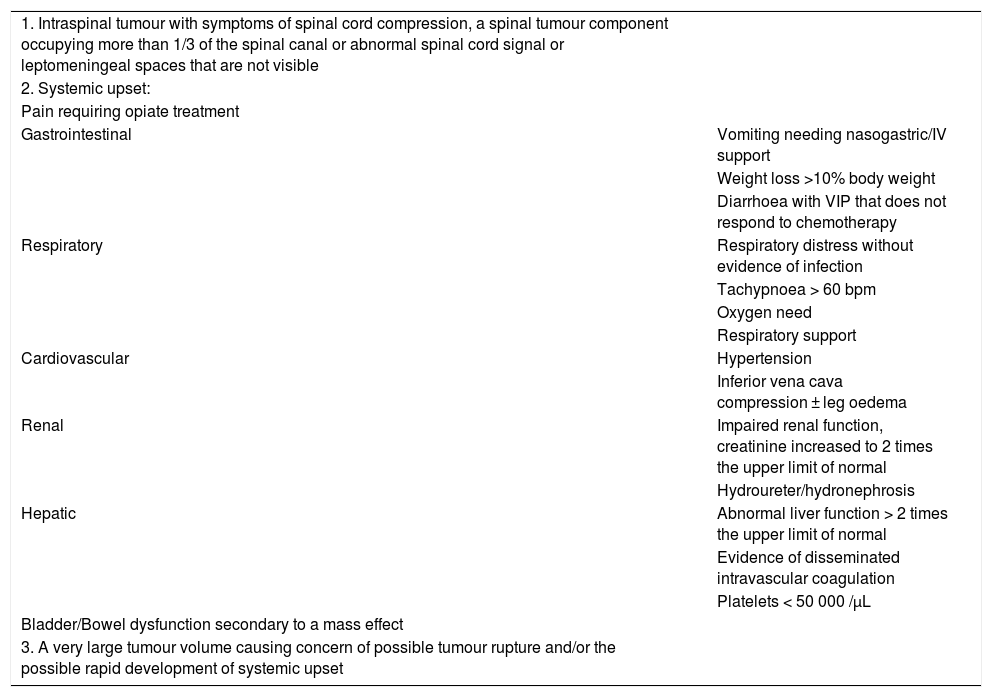
We detailed symptoms classified by the affected organ or system involved and their aggressiveness. In order to use a homogeneous classification of the symptoms of different tumours, we decided to categorise the presentation at onset as “life-threatening symptoms” (LTSs) if the pertinent criteria were met.7 This classification, presented in Table 1, is usually applied to neuroblastoma and is included in the protocols of the International Society of Paediatric Oncology (SIOP).
- d)
Location of primary tumour (qualitative).
- e)
Extension/metastasis (qualitative).
- f)
Histology/cell morphology (qualitative).
- g)
Genetic changes (qualitative).
- h)
Staging based on different protocols applied according to the type of tumour (Table 2) (quantitative).
Table 2.Protocols used for classification of tumours and stages considered “advanced”.
Type of cancer Protocol/Staging system Advanced stage Leukaemia a) 0-12 months LAL-LACTANTES SHOP-2002 protocol High risk or very high risk b) 12-18 months LAL/SEHOP PETHEMA 2013 protocol High-risk Neuroblastoma International Neuroblastoma Risk Group Staging System Metastatic, M (excludes MS) Retinoblastoma Reese-Ellsworth classification Stage III, IV or V CNS tumours Localized or disseminated Disseminated Wilms tumour SIOP UMBRELLA protocol Stage IV or V Hepatoblastoma PRETEXT classification Stage IV or metastatic Sarcoma Non-metastatic RMS Non-metastatic EpSSG RMS-2005 protocol High risk or very high risk Metastatic RMS IRSG protocol Metastatic Ewing sarcoma Localized or metastatic ALL, acute lymphoblastic leukaemia; CNS, central nervous system; EpSSG: European paediatric Soft tissue sarcoma Study Group; IRSG, Intergroup Rhabdomyosarcoma Group; PETHEMA, Programa Español de Tratamientos de Hematología; RMS, rhabdomyosarcoma; SHOP, Sociedad Oncohematología Pediátrica; SIOP: International Society Paediatric Oncology.
- i)
We applied a uniform staging classification by categorising tumours into “not advanced” and “advanced” based on the criteria shown in Table 2.
Life-threatening symptoms.
| 1. Intraspinal tumour with symptoms of spinal cord compression, a spinal tumour component occupying more than 1/3 of the spinal canal or abnormal spinal cord signal or leptomeningeal spaces that are not visible | |
| 2. Systemic upset: | |
| Pain requiring opiate treatment | |
| Gastrointestinal | Vomiting needing nasogastric/IV support |
| Weight loss >10% body weight | |
| Diarrhoea with VIP that does not respond to chemotherapy | |
| Respiratory | Respiratory distress without evidence of infection |
| Tachypnoea > 60 bpm | |
| Oxygen need | |
| Respiratory support | |
| Cardiovascular | Hypertension |
| Inferior vena cava compression ± leg oedema | |
| Renal | Impaired renal function, creatinine increased to 2 times the upper limit of normal |
| Hydroureter/hydronephrosis | |
| Hepatic | Abnormal liver function > 2 times the upper limit of normal |
| Evidence of disseminated intravascular coagulation | |
| Platelets < 50 000 /µL | |
| Bladder/Bowel dysfunction secondary to a mass effect | |
| 3. A very large tumour volume causing concern of possible tumour rupture and/or the possible rapid development of systemic upset |
VIP, vasoactive intestinal peptide.
We have expressed qualitative variables as absolute frequencies and percentages, and quantitative variables as median and interquartile range, as the data did not fit a normal distribution (P < .05 in the Kolmogorov-Smirnov test).
The statistical analysis was performed with the Excel® software, and we generated charts using Excel®, Microsoft Office Publisher® and Power Point®.
ResultsSample characteristicsIn the period under study (January 2007 to August 2019), 72 patients received diagnoses of cancer requiring chemotherapy at the Hospital La Fe. Four of these patients had more than 1 tumour, amounting to a total of 76 tumours in this period.
The most frequent type of tumour was leukaemia (21 patients), followed by neuroblastoma (15 patients), CNS tumours (12 patients), retinoblastoma (9 patients, 13 tumours), WT (7 patients), hepatoblastoma (5 patients) and sarcoma (3 patients). Overall, 70.8% of patients had solid tumours (51/72) and 29.2% had leukaemia (21/72).
The median age at diagnosis was 9.5 months, and there was a slight predominance of the male sex in the sample (54.2% male vs 45.8% female).
Clinical variablesThe clinical presentation of cancer varies depending on the type of tumour, as can be seen in Fig. 1.

Thirteen patients (18.1%) were asymptomatic at diagnosis. In 8 patients, the tumour was an incidental finding, and in 5 the reason for seeking medical care was the detection of an asymptomatic mass: 3 patients with neuroblastoma (clinical presentation of 20% of these tumours) and 2 patients with sarcoma. On the other hand, all patients with CNS tumours (12) had symptoms at onset, mainly focal neurologic signs.
In terms of severity, we ought to highlight that 15 patients had onset with LTSs (20.8%), most of them with tumours of the nervous system: 7 cases of CNS tumours (46.6% of patients with LTSs) and 6 cases of CNS neuroblastoma (40% of patients with LTSs). The remaining patients that had LTSs at onset were 2 patients with leukaemia that presented with respiratory distress and oliguria secondary to tumour lysis syndrome, who required admission to the paediatric intensive care unit (PICU).
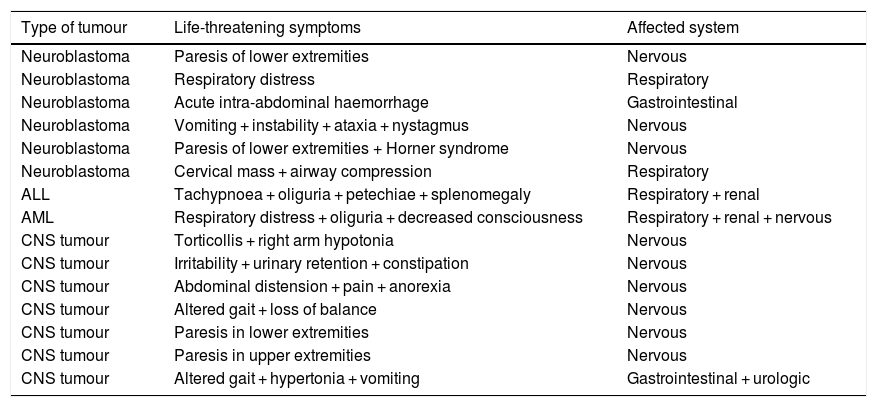
Table 3 presents the distribution of tumours that had onset with LTSs, and Table 4 offers details on specific symptoms observed in the patients (Table 4).
Tumours with life-threatening symptoms at onset and affected system.
| Type of tumour | Life-threatening symptoms | Affected system |
|---|---|---|
| Neuroblastoma | Paresis of lower extremities | Nervous |
| Neuroblastoma | Respiratory distress | Respiratory |
| Neuroblastoma | Acute intra-abdominal haemorrhage | Gastrointestinal |
| Neuroblastoma | Vomiting + instability + ataxia + nystagmus | Nervous |
| Neuroblastoma | Paresis of lower extremities + Horner syndrome | Nervous |
| Neuroblastoma | Cervical mass + airway compression | Respiratory |
| ALL | Tachypnoea + oliguria + petechiae + splenomegaly | Respiratory + renal |
| AML | Respiratory distress + oliguria + decreased consciousness | Respiratory + renal + nervous |
| CNS tumour | Torticollis + right arm hypotonia | Nervous |
| CNS tumour | Irritability + urinary retention + constipation | Nervous |
| CNS tumour | Abdominal distension + pain + anorexia | Nervous |
| CNS tumour | Altered gait + loss of balance | Nervous |
| CNS tumour | Paresis in lower extremities | Nervous |
| CNS tumour | Paresis in upper extremities | Nervous |
| CNS tumour | Altered gait + hypertonia + vomiting | Gastrointestinal + urologic |
ALL, acute lymphoblastic leukaemia; AML, acute myeloid leukaemia; CNS, central nervous system.
In the sample, 45.8% of patients (33/72) had advanced disease at the time of diagnosis (Table 2).
In the subset of patients with solid tumours, 17 (23.6%) had metastases at the time of diagnosis, or 12 (16.6%) of we exclude the 5 cases of MS stage neuroblastoma.
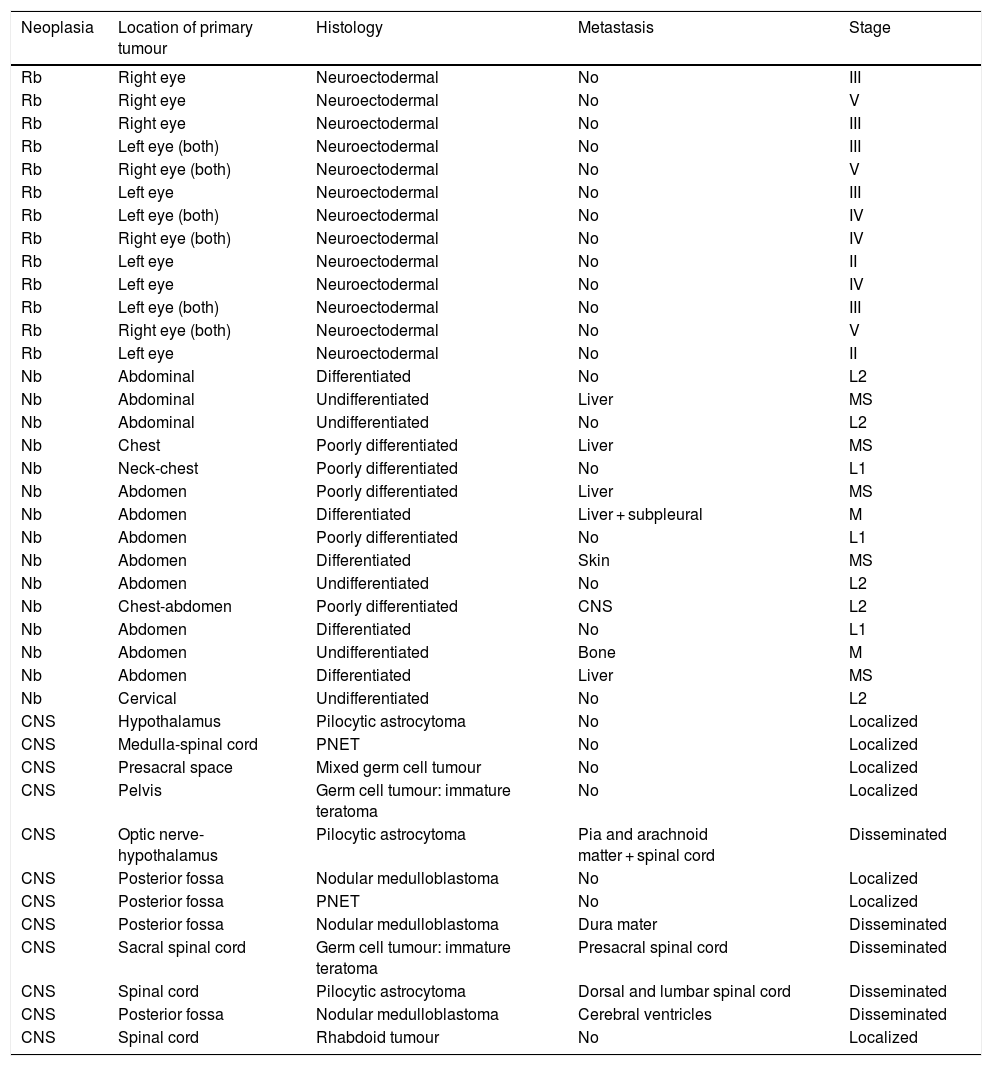
We will now discuss the extension of disease, histological classification and staging of the 3 most prevalent types of solid tumours in the sample: retinoblastoma, neuroblastoma and CNS tumours (Table 5).
Tumour extension, histological classification and staging of retinoblastoma, neuroblastoma CNS tumour cases.
| Neoplasia | Location of primary tumour | Histology | Metastasis | Stage |
|---|---|---|---|---|
| Rb | Right eye | Neuroectodermal | No | III |
| Rb | Right eye | Neuroectodermal | No | V |
| Rb | Right eye | Neuroectodermal | No | III |
| Rb | Left eye (both) | Neuroectodermal | No | III |
| Rb | Right eye (both) | Neuroectodermal | No | V |
| Rb | Left eye | Neuroectodermal | No | III |
| Rb | Left eye (both) | Neuroectodermal | No | IV |
| Rb | Right eye (both) | Neuroectodermal | No | IV |
| Rb | Left eye | Neuroectodermal | No | II |
| Rb | Left eye | Neuroectodermal | No | IV |
| Rb | Left eye (both) | Neuroectodermal | No | III |
| Rb | Right eye (both) | Neuroectodermal | No | V |
| Rb | Left eye | Neuroectodermal | No | II |
| Nb | Abdominal | Differentiated | No | L2 |
| Nb | Abdominal | Undifferentiated | Liver | MS |
| Nb | Abdominal | Undifferentiated | No | L2 |
| Nb | Chest | Poorly differentiated | Liver | MS |
| Nb | Neck-chest | Poorly differentiated | No | L1 |
| Nb | Abdomen | Poorly differentiated | Liver | MS |
| Nb | Abdomen | Differentiated | Liver + subpleural | M |
| Nb | Abdomen | Poorly differentiated | No | L1 |
| Nb | Abdomen | Differentiated | Skin | MS |
| Nb | Abdomen | Undifferentiated | No | L2 |
| Nb | Chest-abdomen | Poorly differentiated | CNS | L2 |
| Nb | Abdomen | Differentiated | No | L1 |
| Nb | Abdomen | Undifferentiated | Bone | M |
| Nb | Abdomen | Differentiated | Liver | MS |
| Nb | Cervical | Undifferentiated | No | L2 |
| CNS | Hypothalamus | Pilocytic astrocytoma | No | Localized |
| CNS | Medulla-spinal cord | PNET | No | Localized |
| CNS | Presacral space | Mixed germ cell tumour | No | Localized |
| CNS | Pelvis | Germ cell tumour: immature teratoma | No | Localized |
| CNS | Optic nerve-hypothalamus | Pilocytic astrocytoma | Pia and arachnoid matter + spinal cord | Disseminated |
| CNS | Posterior fossa | Nodular medulloblastoma | No | Localized |
| CNS | Posterior fossa | PNET | No | Localized |
| CNS | Posterior fossa | Nodular medulloblastoma | Dura mater | Disseminated |
| CNS | Sacral spinal cord | Germ cell tumour: immature teratoma | Presacral spinal cord | Disseminated |
| CNS | Spinal cord | Pilocytic astrocytoma | Dorsal and lumbar spinal cord | Disseminated |
| CNS | Posterior fossa | Nodular medulloblastoma | Cerebral ventricles | Disseminated |
| CNS | Spinal cord | Rhabdoid tumour | No | Localized |
CNS, central nervous system; Nb, neuroblastoma; PNET, primitive neuroectodermal tumour; Rb, retinoblastoma.
Below is a summary of the results regarding specific genetic prognostic markers associated with paediatric cancer:
- 1
Gene fusions involving the KMT2A gene (11q23) in B-cell ALL: Changes present in 46% of cases (6/13), all associated with very high-risk leukaemia.
- 2
NMYC amplification in neuroblastoma8–10: present in 6.6% of cases (1/15). The affected patient had onset with retroperitoneal bleeding caused by tumour rupture that required admission to the PICU. The patient was managed with surgery and chemotherapy per the HR-NEUROBLASTOMA 1.8/SIOPEN protocol, although there were recurrences at 15 months and 5 months after.
- 3
Chromosome abnormalities11,12 were identified in 8 patients (53.3% of cases of neuroblastoma): structural chromosome abnormalities (SCAs) in 3 (37.5%) and numerical chromosome abnormalities (NCAs) in 5 (62.5%).
- 4
RB1 gene changes in retinoblastoma13: In our series, there were 10 patients with retinoblastoma with a total of 13 tumours. Thus, 3 patients had bilateral retinoblastoma (3/10, 30%), all of who were referred for genetic counselling. A c224G > A mutation in exon 2 of the RB1 gene was detected in one of them.
Management with chemotherapy was one of the inclusion criteria. However, this was not the only treatment received by patients, as can be seen in Fig. 2.
Discussion
The diagnosis and treatment of cancer in the first stage of life is a challenge to health professionals that work with paediatric patients. The immaturity of our youngest patients has a significant impact on survival and morbidity, so it is key to be aware of the particular characteristics of this age group to provide adequate management.
In this study, we analysed the experience in our hospital as regards the clinical presentation, diagnosis and treatment of patients given a diagnosis of cancer before age 18 months managed with chemotherapy. To this end, we conducted a retrospective review of the cases managed in our unit over a period of more than 12 years (January 2007 to August 2019).
General characteristicsThe distribution by type of tumour was similar to distributions described in the previous literature, and leukaemia was the most frequent type. We ought to mention that the median age of patients with a leukaemia diagnosis was 11 months (IQR, 9), which was greater compared to the 8.5 months (IQR, 8) of patients with embryonal tumours (neuroblastoma, CNS embryonal tumours, WT, hepatoblastoma and retinoblastoma), a finding that supported the hypothesis that embryonal tumours are most prevalent in the early months of life.
The distribution by sex was also consistent with the previous literature, with a mild predominance of the male sex (54.2%) versus the female sex (45.8%).
Our findings evince the broad range of symptoms found in these patients (Fig. 1), cases of multisystem involvement (Table 4) and the high frequency of aggressive forms:
- a)
In our sample, 20.8% of patients met the criteria for LTSs, a higher proportion compared to other paediatric age groups. The most aggressive tumours were those involving the CNS based on the following findings: first, CNS tumours accounted for 46.6% of cases with LTSs (a proportion that rose to 86.6% if we included all tumours of the nervous system, that is, CNS tumours and neuroblastomas). Second, this was the only type of tumour of which none of the cases were asymptomatic. Third, the symptoms associated most frequently with these tumours were focal neurologic signs, such as hemiparesis, hypertonia, diplopia, urinary retention… Another focal sign that merits comment on account on its misleading “harmlessness” is torticollis, which is the presenting symptom in some brain tumours in infants.
- b)
In contrast, in a large group of patients the diagnosis resulted from the detection of an asymptomatic mass (in 5 cases) or an incidental finding (8 patients).
We now proceed to discuss relevant findings for each type of tumour.
Leukaemia14Based on the existing literature, while lymphoid leukaemia continues to be more prevalent in the early stages of life, the proportion typically declines in favour of myeloid or mixed phenotype leukaemias.15 Indeed, in our sample there were 14 patients with lymphoid leukaemia (14/21 compared to 80% at other ages), 6 with myeloid leukaemia (6/21 compared to 20% in the larger paediatric population) and 1 case of mixed leukaemia (1/21). If we were to consider only the infants in our study (age < 12 months), the results would differ even more compared to the general paediatric population: 7/14 with ALL, 7/14 with AML and 1/14 with mixed phenotype acute leukaemia.
We found gene fusions involving the KMT2A gene (11q23) in 46% of the sample, which was consistent with the literature16 and considerably more frequent compared to other populations.
Central nervous system tumoursCentral nervous system tumours are frequent in the paediatric age group. Although they are usually infratentorial, supratentorial locations predominate in infants.17 This probably accounts for the high proportion of patients presenting with focal neurological signs in this age group, contrary to the findings in other paediatric age groups (with a higher prevalence of manifestations secondary to hydrocephalus). The most frequent histological classification is embryonal. Younger age is a predictor of poor outcomes in this type of tumours.18
In our sample, CNS tumours were third in frequency and with a clear predominance of embryonal tumours (66.6%).
The most frequent location was the spinal cord (6 cases), followed by the posterior fossa (4 cases) and the supratentorial region (2 cases). This atypical distribution could be explained by the fact that the sample consisted of patients managed with chemotherapy, and therefore excluded tumours resistant to chemotherapy or in which curative surgery was the sole treatment. Patients managed with radiotherapy did not start this treatment until at least age 3 years.
As noted before, CNS tumours were the most aggressive tumours in our sample.
NeuroblastomaNeuroblastoma was the second most frequent type of tumour (15 cases), following closely after leukaemia (21 cases). It should be taken into account that some neuroblastomas are treated exclusively with surgery or even with watchful waiting, and since our sample only included patients treated with chemotherapy, our study probably underestimated the actual incidence of neuroblastoma in this age group.
The most frequent location of neuroblastoma is the abdomen, with a relative increase in frequency of tumours in the neck or thorax in infants. In our sample, 73.3% of neuroblastomas were abdominal, while there were 3 cases where it was located in the thorax and 2 cases where it was located in the neck.
The clinical presentation was heterogeneous: we found that 20% of cases were diagnosed after detection of an asymptomatic mass or an incidental finding, while manifestations of disseminated disease were also not infrequent. In 26.6% of cases the patient had focal neurologic signs at onset (paresis of the lower extremities, Horner syndrome or anisocoria) secondary to neural tissue infiltration, while in 6.6% the warning sign was a subcutaneous inguinal mass secondary to cutaneous infiltration.
When it came to the stage of neuroblastoma, the distribution in our sample was as follows: L1, 13.3%; L2, 33.3%; M, 20% and MS, 33.3%.
The MS stage is characteristic of children aged less than 18 months. In the absence of genetic factors associated with a poor outcome, the prognosis of these tumours is good despite infiltration of the liver, spleen or bone marrow, even without treatment.19 In our sample, there were 5 cases of MS stage neuroblastoma (33.3%). The rationale to exclude the watchful waiting approach was the presence of SCAs in 3 patients and the presence of LTSs in 1 patient. The remaining patient had been diagnosed and treated in 2008, when there was insufficient evidence supporting watchful waiting as a possible approach.
RetinoblastomaRetinoblastoma was the fourth most frequent type of tumour in our sample.
Ninety-two percent of these patients (12/13) had leukocoria at onset, a clinical sign that is very sensitive and specific in the diagnosis of retinoblastoma. In this regard, the importance of testing the pupillary light response during routine paediatric checkups cannot be overstated.
Development of retinoblastoma in infants should alert clinicians to the possibility that may be indicative of cancer that is hereditary or carries a germline mutation, more aggressive disease and greater extension. In such cases, it is important to refer the patient to genetic counselling. In our sample, we found a high proportion of patients with bilateral tumours (46.1%), and of patients that required aggressive treatment (38.5% underwent enucleation).
Wilms tumourThe prognosis of WT in infants is more favourable compared to other age groups, as they are frequently epithelial and detected in the early stages.20
In our study, the 7 cases of WT were all epithelial type (100%), and 3 were advanced at the time of diagnosis.
HepatoblastomaThe same can be said of hepatoblastoma in infancy and early childhood: it tends to have good outcomes due to a favourable histology.
In our sample, the histological classification of most cases of hepatoblastoma was epithelial. There was one mixed type case, corresponding to the only patient that had lung metastases at the time of diagnosis.
We ought to highlight that hepatoblastoma is frequently diagnosed as the result of an incidental finding (40%-50% of cases).
SarcomaAlthough extremely rare in this age group, the most frequent sarcoma in infancy or early childhood is rhabdomyosarcoma. In our sample, there was one case of orbital rhabdomyosarcoma and 1 case of rhabdomyosarcoma in the lower extremity (right gastrocnemius). The histology is usually favourable, and the 2 cases in our study were embryonal tumours.
In conclusion, tumours in patients aged less than 18 months differ significantly from tumours in older children in terms of their clinical course, diagnosis and treatment.21 Taking these particularities into account allows early diagnosis, a more appropriate treatment and improved outcomes, and therefore knowledge of these aspects is essential in health care professionals that work with the paediatric population.
In addition, given the increased vulnerability of these patients to anticancer treatments, there is considerable interest in the application of emerging fields in personalised healthcare, such as pharmacogenetics and pharmacogenomics,22–24 which allow optimization of the therapeutic effect of treatments, maximising effectiveness and minimising risks.25
FundingThis study was partially funded by a 2016 research grant of the Fundación Mutua Madrileña, the Asociación Pablo Ugarte and the Asociación Esperanza y Sonrisas.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Urtasun Erburu A, Herrero Cervera MJ, Cañete Nieto A. Cáncer en los primeros 18 meses de vida. An Pediatr (Barc). 2020;93:358–366.
