



La distrofia de Emery-Dreifuss (DED) es una entidad infrecuente, 1/400.000 recién nacidos vivos (prevalencia), asociada a muerte súbita en las primeras décadas de la vida. Es característica la triada clínica de contracturas articulares (codos, cuello y tendón de Aquiles, fundamentalmente), distrofia muscular (debilidad progresiva de musculatura escapulohumeral y peronea) y cardiopatía (arritmias auriculares, trastornos de la conducción y miocardiopatía)1.
La afectación cardiaca aparece en la 2.ª-3.ª década de la vida en forma de arritmias auriculares (flutter, fibrilación, aurícula silente) y bloqueo auriculoventricular, y condiciona el pronóstico1,2.
Presentamos el caso de un varón de 13 años previamente asintomático que consulta por dolor torácico punzante, no irradiado, intenso e intermitente en lado derecho, en reposo, no con ejercicio, sin otros síntomas. Su abuela materna, portadora de marcapasos, estaba intervenida de valvuloplastia tricúspide y mitral. Además, un tío materno afecto de una enfermedad muscular, que no sabían precisar, había sufrido una muerte súbita con 35 años durante el sueño.
El paciente estaba eupneico y bradicárdico, con un soplo II/VI en foco tricuspídeo. En la exploración neurológica destacaba retracción aquílea bilateral leve, de isquiotibiales y de codo derecho, sin debilidad.
El ecocardiograma mostraba importante dilatación de la aurícula derecha (AD) con insuficiencia tricúspide (IT) grave que permitía estimar valores de hipertensión pulmonar moderada (figs. 1A y B). Se realizó un electrocardiograma evidenciando ritmo nodal a 42lpm, sin actividad auricular, así como el eje desviado hacia la derecha (fig. 2, ergometría basal).
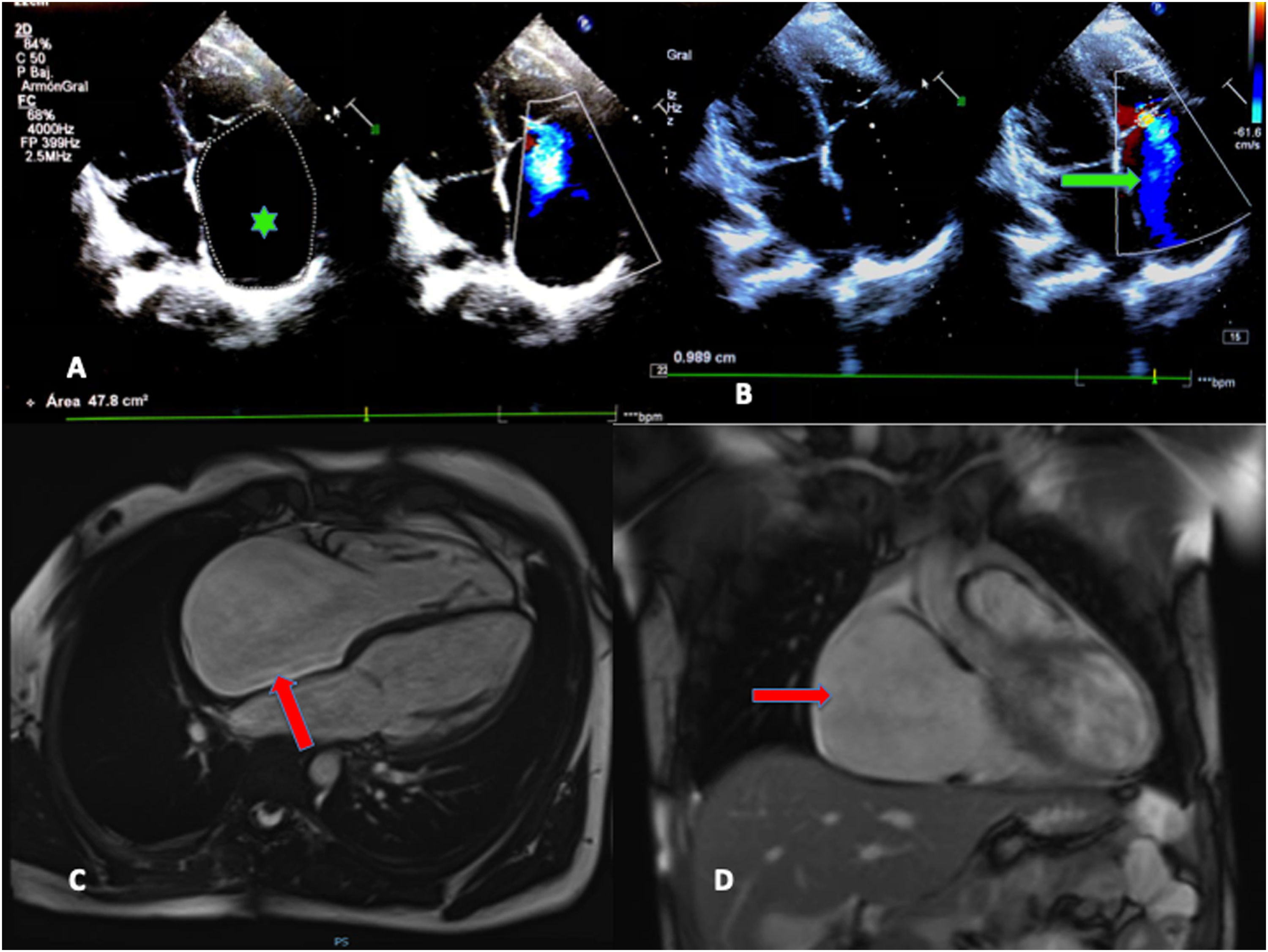
A) Ecocardiograma doppler. Proyección 4 cámaras. Destaca dilatación de la aurícula derecha (AD) (Área por planimetría 2D de 47,8cm2; valores medios para un adulto: 11,43cm2) (asterisco verde). B) Se visualiza flujo de insuficiencia triscuspídea grave con jet que llega hasta el techo de la AD (flecha verde). C) Resonancia magnética. Dilatación importante de la AD en proyección 4 cámaras y en proyección frontal (flechas rojas). No datos de realce tardío ni de trombos intracavitarios.
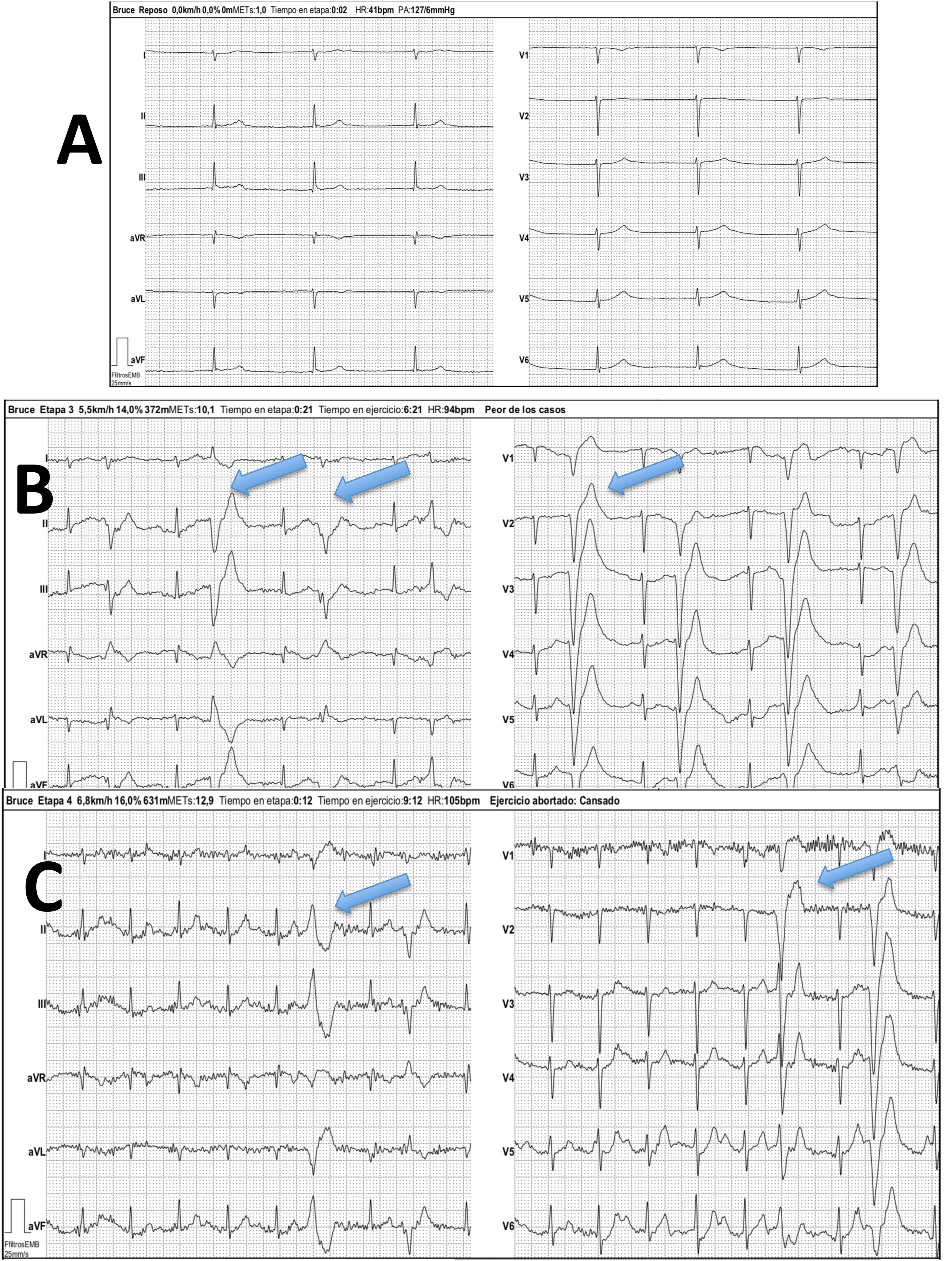
Ergometría según protocolo de Bruce. A) Etapa basal con ritmo de la unión a 42 latidos por minuto (lpm). No actividad auricular. B) Etapa inicial de esfuerzo. Extrasístoles ventriculares polimórficas frecuentes (flechas azules). C) Etapa de esfuerzo máximo con frecuencia cardiaca en torno a 100lpm (50% del teórico predicho) con persistencia de extrasistolia ventricular frecuente (flechas azules).
La resonancia cardiaca confirmó la importante dilatación de la AD, sin imágenes de fibrosis en realce tardío, ni trombos (figs. 1C y D).
En la telemetría durante el sueño se evidenciaron pausas de hasta 4,3s, y la ergometría reveló una incompetencia cronotrópica marcada apareciendo extrasistolia ventricular polimórfica precoz que no desaparecía con el esfuerzo (fig. 2).
La bioquímica sanguínea, incluida creatininquinasa era anodina, sin embargo, el electromiograma mostraba un patrón muscular mixto de afectación peronea.
Se realizó estudio hemodinámico y se midieron presiones confirmando una hipertensión pulmonar mixta con aumento de la presión de arteria pulmonar y de la presión de enclavamiento capilar (16mmHg) (afectación de cavidades izquierdas).
Finalmente, el estudio electrofisiológico mostraba un ritmo de la unión, sin conseguir captura en ninguna región de la AD (aurícula silente, sin actividad eléctrica). El protocolo de inducción de arritmias ventriculares mediante trenes de hasta 3 extra-estímulos fue negativo.
Ante los antecedentes familiares y las retracciones articulares, se planteó un estudio de secuenciación masiva dirigido a enfermedades musculares que asocian trastornos del ritmo cardiaco. Se encontró una variante probablemente patogénica c.266-1G>C en el gen EMD en hemicigosis asociada a distrofia muscular de Duchenne (DED) tipo I ligada al X, que posteriormente se confirmó en la madre.
La DED es una enfermedad muscular de base genética cuyas manifestaciones cardiacas no suelen aparecer en la infancia1,3. Existen fundamentalmente 2 patrones de enfermedad:
- -
La forma de herencia autosómica dominante (gen LMNA, DED tipo 2), la más prevalente, en la que la proteína afectada es la lámina A/C y las arritmias ventriculares son frecuentes junto con anomalías de la conducción auricular.
- -
La forma de herencia ligada al cromosoma X (gen afecto EMD), que fue la primera descrita en 19664. La proteína afectada es la emerina que forma parte de la membrana nuclear interna, y tiene alta expresión en los discos intercalados del músculo cardiaco1–3. La modificación de esta produce alteraciones celulares que conducen al reemplazo del miocardio por tejido adiposo-fibroso con la consecuente dilatación y disfunción de las estructuras afectas.
Típicamente se inicia en las aurículas y luego progresa a nodo AV, y puede afectar a los ventrículos produciendo dilatación y disfunción grave. Hasta el 60-80% de los casos precisan marcapasos entre la 2.ª-3.ª década de la vida y casi el 50% pueden desarrollar posteriormente aurícula silente2,3,5.
La variabilidad en la expresión clínica y la escasez de ensayos controlados hace difícil establecer unas recomendaciones generales, basándose el manejo de estos pacientes en opiniones de expertos.
En este paciente destaca la precocidad y la gravedad de la manifestación cardiaca, presentando un electrocardiograma muy patológico, clave para el enfoque del caso.
Habitualmente la afectación cardiaca suele presentarse tras las manifestaciones musculoarticulares, habiendo pasado inadvertidas las retracciones articulares en nuestro caso.
Ante un caso con trastorno de la conducción se debe considerar esta entidad e indagar sobre antecedentes familiares y la sintomatología muscular que, como en este adolescente, puede pasar desapercibida. Aunque no existe un tratamiento específico, la colocación de un marcapasos o incluso un desfibrilador, puede salvar la vida de estos pacientes, por lo que es fundamental realizar un diagnóstico precoz.
