



Emery-Dreifuss muscular dystrophy (EDMD) is a rare disease with an incidence of 1 per 400 000 live births associated with sudden death in the first decades of life. It is characterised by the triad of joint contractures (chiefly elbows, neck and Achilles tendon), muscular dystrophy (progressive wasting of scapulohumeral and peroneal muscles) and heart disease (atrial arrhythmias, conduction disorders and cardiomyopathy).1
Cardiac involvement usually manifests in the second or third decade of life in the form of atrial arrhythmias (flutter, fibrillation, atrial standstill) and atrioventricular block, and determines the prognosis.1,2
We present the case of a boy aged 13 years who was previously asymptomatic and presented with sharp, stabbing, nonradiating, intense and intermittent right-sided chest pain at rest, not triggered by physical activity, with no other symptoms. His maternal grandmother, who carried a pacemaker, had undergone tricuspid and mitral valvuloplasty. In addition, a maternal uncle who suffered from a muscular disorder that had not been identified precisely had died suddenly at age 35 years in his sleep.
The patient had normal breathing and bradycardia, with detection of a grade II/VI murmur best heard at the level of the tricuspid valve. The salient findings of the neurologic examination were mild contracture of the Achilles tendon and hamstrings on both sides and the right elbow, with no muscular weakness.
The echocardiogram revealed substantial enlargement of the right atrium (RA) and severe tricuspid regurgitation and allowed the estimation of values indicative of moderate pulmonary hypertension (Fig. 1, A and B). A subsequent electrocardiogram showed junctional rhythm at a rate of 42 beats per minute (bpm), absence of atrial activity and right axis deviation (Fig. 2, baseline stress test tracings).
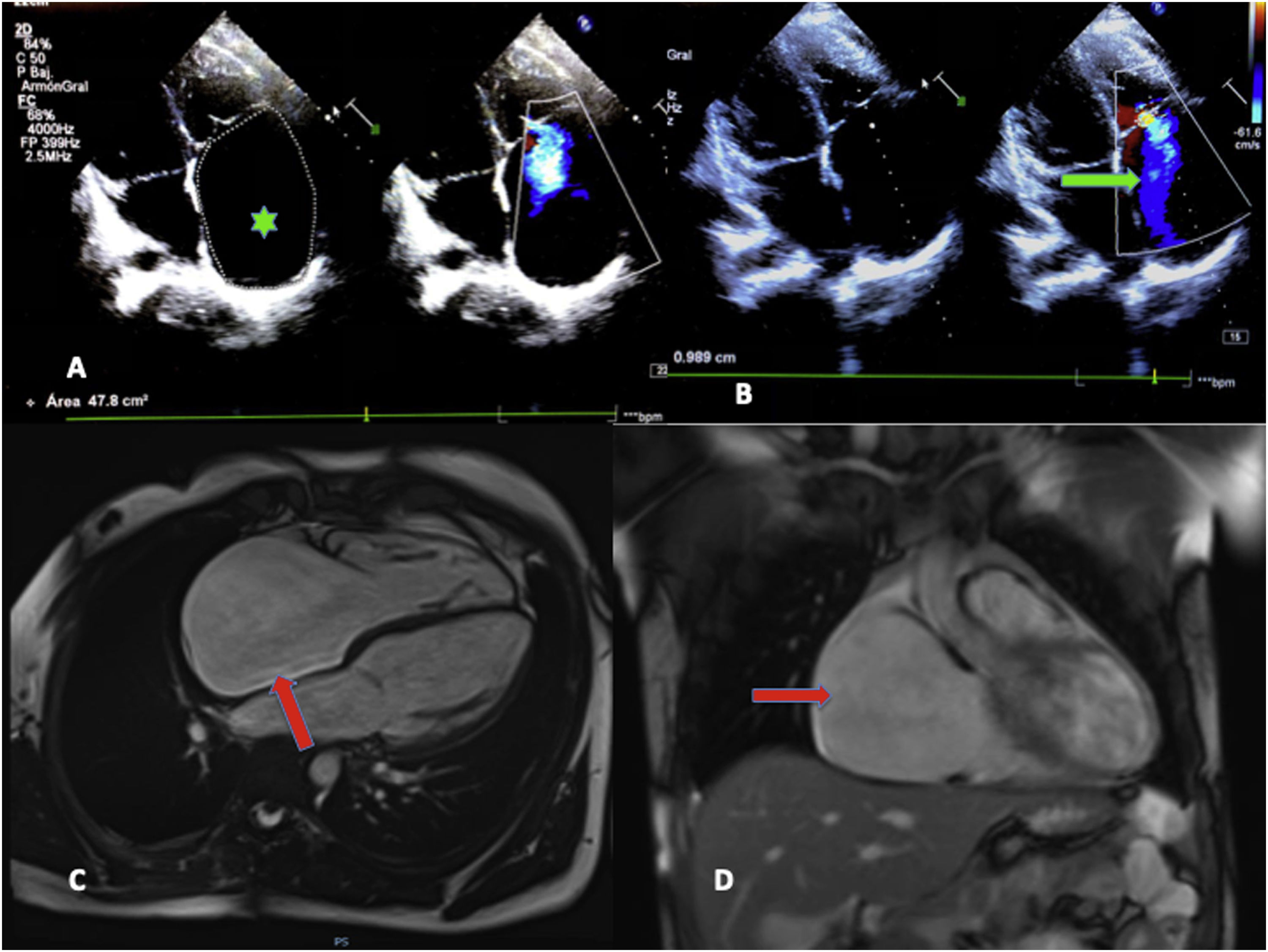
A) Doppler echocardiogram. Four-chamber view. The chief finding was the dilation of the right atrium (RA) (2D planimetry area, 47.8 cm2; mean value for an adult, 11.43 cm2) (green asterisk). B) Severe tricuspid insufficiency, with visualization of regurgitation jet reaching the dome of the RA (green arrow). C) Magnetic resonance imaging. Significant dilation of the RA in the 4-chamber and coronal views (red arrows). No delayed enhancement or evidence of intracardiac thrombi.
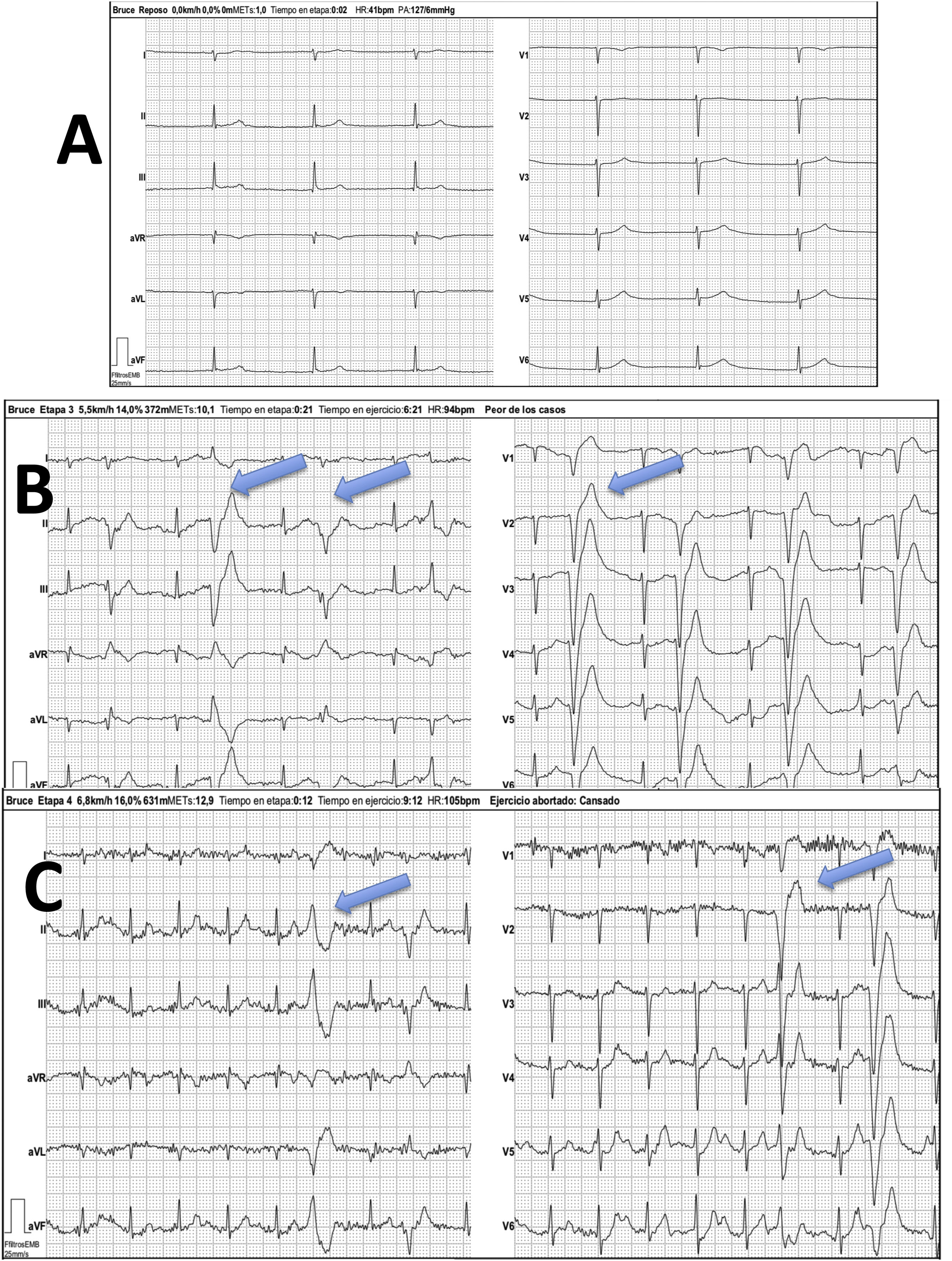
Bruce protocol treadmill test. A) Baseline stage with junctional rhythm at a rate of 42 bpm. Absence of atrial activity. B) Initial stage, frequent polymorphic ventricular asystole (blue arrows). C) Maximum stress stage, with heart rate at approximately 100 bpm (50% of predicted value) with persistence of frequent ventricular asystole (blue arrows).
The cardiac MRI confirmed the significant enlargement of the RA, without evidence of fibrosis or thrombi in late enhancement imaging (Fig. 1, C and D).
The asleep electroencephalogram evinced pauses lasting up to 4.3 s and the cardiac stress test evinced marked chronotropic incompetence with early development of polymorphic ventricular extrasystoles that did not disappear with physical activity (Fig. 2).
The blood chemistry panel was unremarkable, including the level of creatine kinase, but the electromyogram evinced a mixed pattern of muscle involvement in the peroneal region.
The patient underwent a haemodynamic assessment with measurement of blood pressures that confirmed the presence of combined pre- and post-capillary pulmonary hypertension with increased pulmonary artery and pulmonary capillary wedge pressures (16 mmHg) (left heart involvement).
Lastly, the electrophysiological study revealed junctional rhythm, with no atrial capture achieved anywhere in the RA (atrial standstill, absence of electrical activity). The results of the ventricular arrhythmia induction protocol with trains with up to 3 extrastimuli were negative.
Given the family history and the presence of joint contractures, we ordered a next generation sequencing panel that targeted muscular diseases associated with heart rhythm. It identified a hemizygous likely pathogenic variant in the EMD gene (c.266-1G>C) associated with type 1 X-linked Duchenne muscular dystrophy (EDMD), whose presence was subsequently confirmed in the mother.
Emery-Dreifuss muscular dystrophy is a genetic muscle disorder in which cardiac manifestations usually appear after childhood.1,3 There are 2 main forms of disease:
- -
The form with autosomal dominant inheritance (LMNA gene, EDMD type 2), the most prevalent one, in which the affected protein is lamin A/C, frequently manifesting with frequent ventricular arrhythmias and atrial conduction disorders.
- -
The X-linked form (involving the EMD gene), which was described first in 1966.4 The affected protein is emerin, which is found in the inner nuclear membrane and highly expressed in the intercalated discs of cardiac muscle.1–3 Changes in this protein cause cellular abnormalities leading to the replacement of myocardial tissue by adipose-fibrous tissue, which in turn result in the dilation and dysfunction of affected structures.
Cardiac involvement typically starts in the atria, progresses to the atrioventricular (AV) node and may eventually involve the ventricles, causing severe enlargement and dysfunction. Up to 60% to 80% of affected patients need a pacemaker by the second or third decade of life, and up to nearly 50% may develop atrial standstill.2,3,5
The heterogeneity of the clinical presentation of EDMD and the lack of randomised controlled trials precludes the development of general recommendations, so the management of affected patients is based on expert opinions.
The salient characteristics of this case were the early development and severity of cardiac manifestations, with significant abnormalities in the ECG that guided the management of the patient.
Cardiac involvement usually develops after the musculoarticular manifestations, but in the case presented here, the articular contractures had gone undetected.
This disease should be contemplated in patients with conduction disorders, investigating the family history and assessing for muscular manifestations that, as was the case of our patient, may have not been noticed. Although there is no specific treatment for EDMD, placement of a pacemaker or even a defibrillator may be lifesaving, and therefore early diagnosis is crucial.
