

Sr. Editor:
La ataxia-telangiectasia (AT) es una entidad hereditaria, caracterizada, en sus formas completas, por un cuadro de ataxia cerebelosa progresiva, telangiectasias en piel y conjuntivas, defectos inmunológicos (usualmente una inmunodeficiencia combinada que se traduce en la aparición de infecciones sinopulmonares recurrentes) y una mayor tendencia al desarrollo de tumores malignos por hipersensibilidad a las radiaciones ionizantes1-3.
La siringomielia es un trastorno degenerativo esporádico, consistente en la presencia de una cavidad en el interior de la médula espinal o el bulbo. Desde el punto de vista clínico cursa con debilidad muscular y trastornos de la sensibilidad (anestesia termoalgésica segmentaria con preservación de las sensibilidades táctil y propioceptiva).
Presentamos un caso de siringomielia en un paciente diagnosticado previamente de AT.
Varón de 8 años de edad, diagnosticado con 18 meses de AT. Es seguido en nuestras unidades de neuropediatría y de inmunodeficiencias. En control evolutivo refiere la aparición de cuadro de dolor a nivel de extremidades superiores y espalda, junto con un empeoramiento llamativo de los trastornos de la marcha y aumento de la inestabilidad.
Antecedentes personales: Otitis e infecciones de vías respiratorias altas frecuentes. Neumonía en una ocasión. Tratamiento con inmunoglobulinas intravenosas desde los 6 años de edad.
Desarrollo psicomotor: sedestación a los 7 meses. Marcha liberada a los 12 meses. Bisílabos: 17 meses.
Conexión con el medio, aprendizaje y cognitivo normales. A los 20 meses comienza a manifestar marcha inestable y telangiectasias conjuntivales.
Antecedentes familiares: Tíos y primos maternos con sintomatología similar a la del paciente, fallecidos a la edad de 10-12 años.
Exploración física: Constitución asténica. Piel: telangiectasia en conjuntivas, no presentes en otras localizaciones. Exploración neurológica: hallazgos destacables: apraxia oculomotora; ataxia troncular y dismetría en extremidades. Movimientos atetósicos de extremidades superiores ocasionales. Arreflexia e hipotonía muscular de las 4 extremidades. Exploración de sensibilidad profunda y superficial normal.
Pruebas complementarias: Alfa-fetoproteína 205,5 ng/ml. Inmunoglobulinas séricas: IgG 490 mg/dl, IgA 171 mg/dl, IgM 320 mg/dl. Subpoblaciones linfocitarias: CD3 645/μl (39,4 %), CD4 242/μl (38 %), CD8 210/μl (32 %), CD19 93/μl (14,4 %), CD16-CD56 247/μl (38 %).
RM craneal: atrofia cerebelosa con leve aumento de IV ventrículo. Resto, normal. RM espinal: dilatación hidrosiringomiélica en médula dorsal y lumbar (fig. 1).
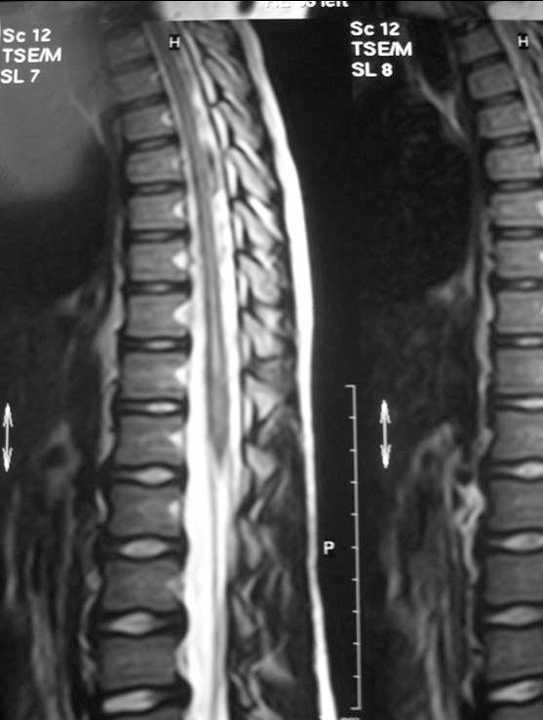
Figura 1.RM axial que muestra dilatación siringomiélica a nivel dorsolumbar.
La AT es una enfermedad rara, que se hereda de un modo autosómico recesivo. Se estima una incidencia en torno a 1/80.000-100.000 nacidos vivos4. Es causada por una mutación en el gen ATM (locus 11q.22.3) que parece implicado en la síntesis de una proteína con actividad fosfoinositol-3 cinasa implicada en la regulación del ciclo celular y la reparación del ADN5,6.
La siringomielia a menudo se asocia a malformaciones congénitas de la unión craneovertebral, como la malformación de Arnold-Chiari u otras alteraciones, la escoliosis torácica, fusión de vértebras o anomalía de Klippel-Feil. Los casos de siringomielia adquiridos se deben a aracnoiditis basal, o pueden desarrollarse en un segmento de médula de forma secundaria a tumores intramedulares, aracnoiditis espinal o traumatismo grave. Desde el punto de vista clínico cursa con debilidad muscular y trastornos de la sensibilidad (anestesia termoalgésica segmentaria con preservación de las sensibilidades táctil y propioceptiva).
La aparición en pacientes con AT de otros signos neurológicos o rápida progresión de los síntomas deben hacernos sospechar la posibilidad de tumores, infecciones o malformaciones asociadas. En este caso, el dolor y la rápida progresión de las alteraciones en la marcha indicaron el examen neurorradiológico del neuroeje, lo cual finalmente condujo al diagnóstico de siringomielia. La ausencia de síntomas clásicos puede ser debida a un diagnóstico precoz o a alteraciones neurosensoriales propias de la enfermedad de base.
La afectación neurológica en pacientes con AT suele iniciarse en torno a los 12-18 meses, y consiste en alteraciones de la marcha que desembocan en un cuadro cerebeloso lentamente progresivo, debido a la desaparición gradual de las células de Purkinje.
En la AT se han descrito otros procesos degenerativos a nivel de médula espinal, tronco cerebral y médula espinal7. Estos cambios probablemente son debidos a alteraciones en la regulación del ciclo celular producidos por el producto defectivo del gen ATM8.
En el caso que presentamos, el paciente no padeció en ningún momento los procesos que clásicamente se atribuyen a la etiopatogenia de la siringomielia, por lo que pensamos que puede tratarse de una asociación relacionada con los cambios degenerativos propios de la enfermedad. Aunque la existencia de estos cambios son conocidos, en la literatura médica revisada no hemos encontrado previamente descrita la asociación de AT con siringomielia.
Correspondencia: juanl.santos.sspa@juntadeandalucia.es
