


Caso clínico
Recién nacido varón que ingresa en el servicio de neonatología por bajo peso para la edad gestacional y cianosis perinasobucal con las primeras tomas. Antecedentes obstétricos: madre primigesta nulípara que cursa con un embarazo de 40 semanas de gestación controlado. Oligoamnios y cesárea por alteraciones en la frecuencia cardíaca fetal. Test de Apgar al minuto de 7 y a los 5 min de 8. Precisa reanimación con mascarilla con presión positiva intermitente. Antecedentes familiares: familiar de segundo grado con poliosis y albinismo parcial. Exploración física: peso: 2.260 g (< P10). Escaso panículo adiposo. Megacefalia, piebaldismo, hipertelorismo, raíz nasal ancha con orificios nasales antevertidos. Labio superior fino, comisura hacia abajo y filtro largo. Mechón de cabello blanco (fig. 1). Pabellones auriculares bajos y rotados. Cuello corto. Hipospadias peneano (grado II) (fig. 2). Criptorquidia bilateral. Bolsas escrotales pequeñas. Membranas interdigitales palmares. Auscultación cardiopulmonar: soplo holosistólico en borde paraesternal izquierdo III/VI. Pulsos periféricos presentes y simétricos. Abdomen globuloso, sin visceromegalias ni masas. Evolución: presenta retraso en la evacuación meconial (a las 24 h de vida y tras estimulación con sonda rectal y enema) y a las 36 h de vida comienza con un cuadro de seudoobstrucción intestinal. Pruebas complementarias: enema opaco sugerente de megacolon con segmento agangliónico corto y retraso en la eliminación de contraste. Manometría: ausencia de reflejo de relajación del esfínter anal interno. Biopsia anorrectal: ausencia de células ganglionares en el segmento afectado con aumento de la acetilcolinesterasa y aumento de las terminaciones nerviosas. Potenciales auditivos evocados: sordera neurosensorial bilateral profunda. Estudio cardiológico: comunicación interventricular perimembranosa.
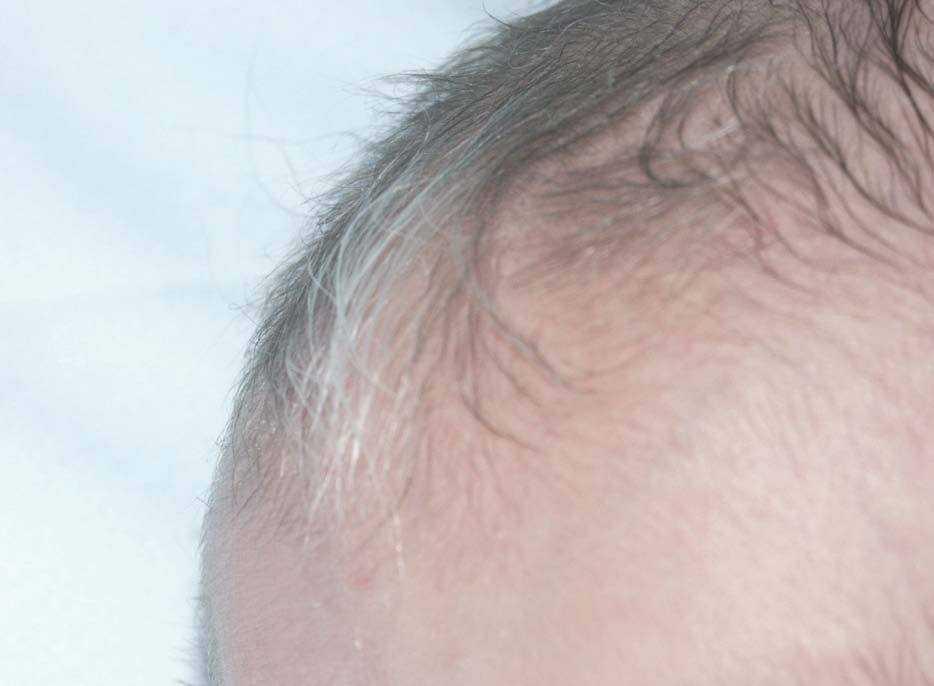
Figura 1. Mechón de cabello blanco frontal.
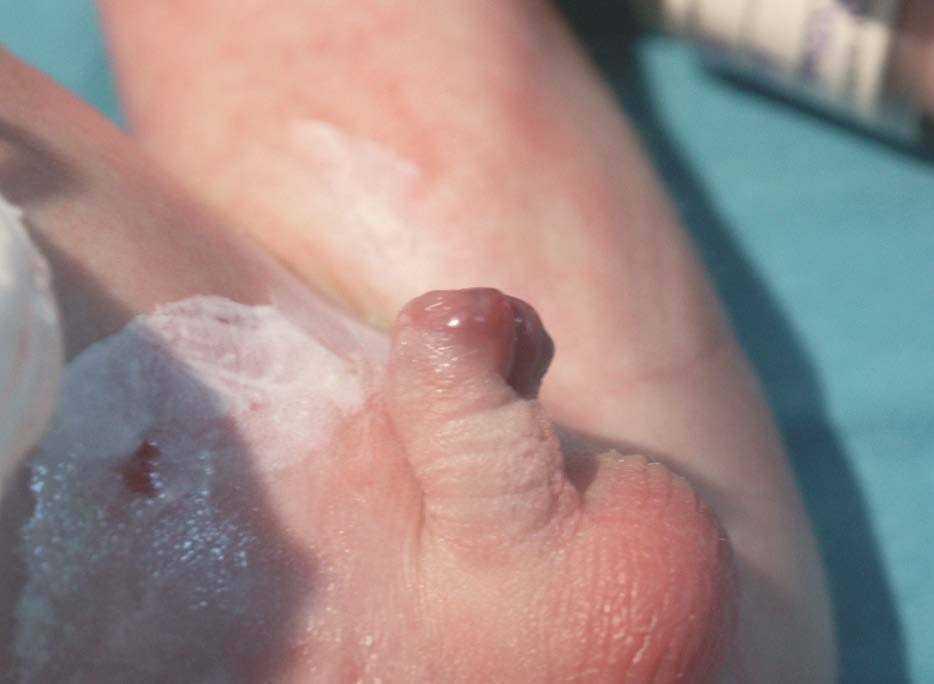
Figura 2. Hipospadias peneano (grado II).
Pregunta
¿Cuál es su diagnóstico?
Síndrome de Waardenburg tipo IV o Waardenburg-Shah (WS)
El síndrome Waardenburg engloba a un grupo de enfermedades hereditarias producidas por una alteración de la migración de las células derivadas de la cresta neural entre la octava y décima semanas de gestación. Células de las que derivan los melanocitos que migran para la formación de la estría vascular del órgano de Corti, la retina, hueso frontal, grupos musculares de miembros superiores, las estructuras palpebrales y para formar las células ganglionares de los plexos submucosos y mientéricos a nivel del tubo digestivo1.
Los criterios diagnósticos del síndrome de Waardenburg son clínicos. Considerándose válido el diagnóstico cuando existen dos criterios mayores o uno mayor y dos menores. Los criterios mayores son: sordera neurosensorial congénita, alteraciones pigmentarias del iris, hipopigmentación del cabello (mechón de cabello blanco), familiar de primer grado afectado y distopia cantorum. Los criterios menores son: manchas cutáneas hipopigmentarias, sinofidria, raíz nasal ancha, orificios nasales antevertidos, poliosis y filtro labial corto2.
Existen 4 variantes de la enfermedad de Waardenburg3:
Tipo I: presenta todas las manifestaciones faciales y visuales que caracterizan el síndrome. El 25 % presentan sordera neurosensorial y la mayoría distopia cantorum.
Tipo II: no presentan distopia cantorum, más del 70 % presentan sordera neurosensorial y el resto de alteraciones faciales y visuales son menos frecuentes4.
Tipo III o Waardenburg-Klein: es similar al tipo I asociando alteraciones musculoesqueléticas y en algunos pacientes retraso mental y microcefalia.
Tipo IV o Waardenburg-Shah: asocia las características del síndrome de Waardenburg a megacolon congénito agangliónico (enfermedad de Hirschsprung)5.
Otras malformaciones que pueden aparecer asociadas a las características de este síndrome son: meningocele, alteraciones cardíacas, atresia anal y malformaciones uterinas y vaginales. En el caso presentado existía una comunicación interventricular, y aunque no hemos encontrado otros casos descritos en la literatura especializada, hipospadias peneana y criptorquidia bilateral.
La incidencia del síndrome de Waardenburg es de 1/270.000 nacidos vivos, aunque debido a su baja prenetrancia (20 %) la frecuencia total estimada es de 1/42.000 personas. Este síndrome es además el causante de hasta el 3 % de los casos de sordera neurosensorial. De los 4 tipos descritos, el tipo IV el más infrecuente existiendo en la literatura especializada unos 50 casos descritos.
Las mutaciones genéticas por las que se producen cada tipo se encuentran en distintos genes y a diferencia de los tipos I, II y III de herencia autosómica dominante, de penetrancia variable. El asociado a enfermedad de Hirschsprung se hereda como un rasgo autosómico recesivo cuando hay mutaciones en los genes EDNRB o EDN3 o como un rasgo autosómico dominante cuando es debido a la mutación del gen SOX106. El síndrome de Waardenburg tipo III puede asociar retraso mental y en el tipo IV sólo algunos pacientes con mutaciones en SOX10 pueden presentar afectación neurológica adicional7.
No existe tratamiento específico para el síndrome de Waardenburg y la mayoría de los pacientes afectados tienen una esperanza de vida normal. Con respecto a la deficiencia auditiva en los tipos I, III y IV suele permanecer estacionaria mientras que en el tipo II puede ser progresiva8. Su diagnóstico precoz es importante para un adecuado desarrollo del lenguaje, educación y los problemas psicológicos que pueda ocasionar. En el tipo III las alteraciones óseas y musculares pueden ser incapacitantes y requerir tratamiento ortopédico y el caso del tipo IV es necesaria la cirugía para la enfermedad de Hirschsprung. Se debe buscar asesoramiento genético para los futuros padres con antecedentes familiares de síndrome de Waardenburg.
Por tanto ante un cuadro de hipopigmentación asociado a una obstrucción intestinal en un recién nacido, habrá que plantearse este diagnóstico sindrómico y realizar un estudio genético del paciente y la familia.
Correspondencia: Dra. A. Abril Molina.
Servicio de Neonatología. Hospital Clínico Universitario San Cecilio.
Cuenca, 2. 18002 Granada. España.
Correo electrónico: anabril15@hotmail.com
Recibido en septiembre de 2005.
Aceptado para su publicación en julio de 2006.
