





Wilms’ tumour is the most frequent renal tumour in children. Multi-modal treatment includes chemotherapy and surgery, with or without radiotherapy. The survival is excellent, with rates exceeding 90%. A review is presented on our experience over the last 15 years of treating Wilms’ tumour in Hospital Niño Jesús, Madrid.
Patients and methodsA retrospective study was conducted on 40 consecutive paediatric patients diagnosed with nephroblastoma between 2002 and 2016 in the Hospital Niño Jesús in Madrid. The clinical characteristics, diagnostic methods, treatment, and follow-up were analysed.
ResultsOf the 40 patients, 23 were boys, with a median age at diagnosis of 2.5 years (range, 4 months–15 years). Three patients underwent initial nephrectomy, three received a fine needle aspiration biopsy, followed by chemotherapy, and 34 patients started pre-operative chemotherapy directly. The median follow-up of the patients was 6.75 years (range, 10 months–13.92 years).
Two patients died from disease progression. There were no treatment-related deaths. Overall survival and event-free survival at 5 years was 94.6±3.7% and 89.4±5%, respectively.
ConclusionWilms’ tumour treatment is a success of modern medicine, currently achieving a survival rate of 95% in our series.
El tumor de Wilms es el tumor renal más frecuente en la edad pediátrica. Su tratamiento es multimodal: incluye quimioterapia y cirugía, con o sin radioterapia. La supervivencia de estos pacientes es excelente, superando el 90%. Presentamos la experiencia de nuestro centro en el tratamiento del tumor de Wilms durante los últimos 15 años.
Pacientes y métodosSe ha realizado un estudio retrospectivo de 40 pacientes pediátricos diagnosticados de forma consecutiva de nefroblastoma entre 2002 y 2016 en el Servicio de Hemato-Oncología pediátrica del Hospital Niño Jesús de Madrid. Se analizaron las características clínicas, los métodos diagnósticos, el tratamiento realizado y la evolución posterior.
ResultadosDe los 40 pacientes, 23 eran niños con una mediana de edad al diagnóstico de 2,5 años (rango, 4 meses-15 años). A 3 pacientes se les realizó nefrectomía inicial, 3 recibieron una punción aspiración con agua fina, seguida de quimioterapia y 34 pacientes recibieron quimioterapia preoperatoria directamente. La mediana de seguimiento de los pacientes fue de 6,75 años (rango, 10 meses-13,92 años).
Dos pacientes fallecieron de progresión de su enfermedad. Ningún paciente falleció de toxicidad en relación con el tratamiento. La supervivencia global y la supervivencia libre de evento a los 5 años fue del 94,6±3,7% y 89,4±5%, respectivamente.
ConclusiónEl tratamiento del tumor de Wilms es un éxito de la medicina moderna, consiguiendo en la actualidad una supervivencia que en nuestra serie alcanza el 95%.
Wilms tumour or nephroblastoma is the second most common intra-abdominal cancer and the fifth most common malignancy in the paediatric age group. It accounts for approximately 6% of all paediatric cancers and is the most frequent tumour of the kidney (more than 95% of all tumours of the kidney in the paediatric age group).1–3 The differential diagnosis of Wilms tumour includes neuroblastoma and other paediatric renal malignancies, such as clear cell sarcoma, malignant rhabdoid tumour and renal cell carcinoma, which have less favourable outcomes.1,4
Patients with several congenital anomalies, such as WAGR syndrome, Denys-Drash syndrome, Beckwith-Wiedemann syndrome, or isolated hemihypertrophy are at higher risk of developing Wilms tumour.3
The incidence of nephroblastoma peaks between ages 2 and 5 years, and 95% of cases are diagnosed before age 10 years.5 The most frequent initial sign is the chance detection of an abdominal mass, followed by haematuria. Some patients may have arterial hypertension due to renal ischaemia, which is caused by the pressure exerted by the tumour on the renal artery. On occasion, patients present with systemic symptoms such as asthenia, anorexia, weight loss and fever.3 Between 10% and 25% may have disseminated disease, usually in the lungs.6
The management of renal tumours has advanced spectacularly since the introduction of multimodal treatment, which includes chemotherapy and surgery with or without radiation. The 5-year survival has increased drastically in the past three decades, from 25% in the pre-chemotherapy era of the late 1960s and early1970s to 90% in the 1990s.3,4,7
Two large collaborative groups have been formed for the investigation of Wilms tumour: the National Wilms Tumor Study Group (NWTSG) in the United States and the International Society for Paediatric Oncology (SIOP) in Europe. These groups disagree on the timing of surgery. The NWTSG recommends upfront nephrectomy for the purpose of histological diagnosis and accurate staging.8–12 The SIOP Renal Tumour Study Group recommends preoperative chemotherapy to shrink the tumour and facilitate surgery—through the prevention of tumour rupture—followed by staging.1,3,8 The overall survival rate is similar for both approaches.11,13,14 Performance of a confirmatory biopsy before chemotherapy in cases of Wilms tumour with typical clinical and radiological characteristics is not routine practice in the SIOP protocols, as evidence from research comparing needle biopsy specimens collected before preoperative chemotherapy and surgical specimens from the subsequent nephrectomy in the same patients found a 95% agreement in the results.13
Given the high survival rate, the main goal at present is to individualise treatment based on the correct risk stratification of patients in order to achieve the highest possible cure rate while minimising the frequency and intensity of acute and late toxicity.5
Based on the correlation between histological characteristics and survival, the SIOP has defined 3 prognostic groups: low risk, intermediate risk and high risk.5,15 The most powerful predictor of an unfavourable outcome is anaplastic histology. Other histological subtypes included in the high-risk group are blastemal histology, clear cell sarcoma and renal rhabdoid tumour.10,13 The second most important prognostic factor is the stage. As is the case in most tumours, the lower the stage the better the prognosis.
Other factors used in the risk stratification of Wilms tumour are patient age, tumour size and response to treatment.16 Based on these factors, patients are classified into one of the 3 risk groups and treated accordingly.2,7
The survival of patients with Wilms tumour is generally excellent, exceeding 90%.2 The rate is 95% for patients with stages I and II Wilms tumour, 75–80% for patients with stage III, and 65–75% for patients with stage IV (Table 1). Only 15% of patients with a favourable histology experience recurrence, compared to an incidence of 50% in those with anaplastic histology. Even patients with recurrent disease have a high survival rate of 60%.7 The most common sites of recurrence are the lungs, pleura, tumour bed and liver. Among patients with metastases, those with liver involvement have a poorer prognosis compared to those with lung metastases.5
Materials and methodsDuring the 15-year study period (January 1, 2002 to June 1, 2016), 40 patients received a diagnosis of nephroblastoma and were managed in the Department of Paediatric Haematology and Oncology of the Hospital Infantil Universitario Niño Jesús. We also included another 2 patients who were referred to our hospital when their cancer relapsed in the analysis of relapsed patients.
We reviewed the health records of the patients to collect data on age, sex, presentation at diagnosis, histology, staging, overall survival and event-free survival. We also analysed the initial treatment, type of recurrence and treatment of recurrence.
The evaluation included a complete blood count, measurement of serum creatinine and electrolyte and liver panels. These tests were performed prior to and during chemotherapy. The evaluation was completed with an abdominal ultrasound and a lung X-ray or CT scan. An abdominal MRI or CT scan was performed in most patients to assess tumour size, staging and operability.
Tumours were classified based on histology and staged according to the SIOP criteria in all patients (Tables 1 and 2).
SIOP staging criteria for renal tumours of childhood (2001).
| Stage I | The tumour is limited to the kidney |
| There is no residual tumour in the resection margins | |
| The vessels of the renal sinus are not involved | |
| Intrarenal vessel involvement, potential involvement of capsule, adjacent tissues, renal sinus vessels and vena cava | |
| Stage II | The tumour extends outside the renal parenchyma and may have infiltrated the capsule, adjacent tissues, renal sinus vessels or vena cava, but it is completely excised |
| Stage III | Incomplete excision: |
| – Involvement of abdominal lymph nodes | |
| – Tumour rupture before or during surgery | |
| – The tumour has penetrated the peritoneal surface | |
| – Tumour thrombi present at resection margins | |
| Haematogenous metastases (lung, liver, bone, …) | |
| Stage IV | Lymph node metastases outside the abdominopelvic region |
| Stage V | Bilateral renal tumours (each side should be substaged separately) |
Revised SIOP classification of renal tumours of childhood (2001).
| Low risk tumours |
| Cystic partially differentiated nephroblastoma |
| Completely necrotic nephroblastoma |
| Intermediate risk tumours |
| Nephroblastoma-epithelial type |
| Nephroblastoma-stromal type |
| Nephroblastoma-mixed type |
| Nephroblastoma-regressive type |
| Nephroblastoma-focal anaplasia |
| High risk tumours |
| Nephroblastoma-blastemal type |
| Nephroblastoma-diffuse anaplasia |
| Clear cell sarcoma of the kidney |
| Rhabdoid tumour of the kidney |
All patients received treatment conforming to the SIOP 2001 nephroblastoma protocol. Thus, patients received preoperative chemotherapy followed by surgery, except 3 patients in whom nephrectomy was performed immediately after diagnosis. A core needle biopsy was performed in 3 patients before initiating treatment (as excisional biopsy is recommended against due to the risk of upstaging) on account of atypical clinical or radiological findings. In all 3 cases, nephroblastoma was confirmed by histological examination of the surgical specimen.
Patients with localised disease received standard chemotherapy with 4 weeks of actinomycin D and vincristine. Patients with advanced disease received 6 weeks of 3-drug chemotherapy with actinomycin D, vincristine and doxorubicin. After completing initial treatment, patients underwent a CT or MRI scan to assess the response of the tumour and plan the surgery. Tumour response was evaluated based on the reduction in the maximum diameter of the tumour from observed in the post-chemotherapy scan compared to the pre-chemotherapy scan.
When it came to postoperative treatment, 3 patients with low-risk stage I disease required no further treatment. The rest received postoperative chemotherapy for 4–34 weeks. The chemotherapy regimen was based on tumour staging and histology.
Patients with tumours limited to the kidney with favourable histology received actinomycin D and vincristine for 4 weeks. Patients with more advanced disease received actinomycin D and vincristine with or without doxorubicin for 27 weeks. Patients with unfavourable histology received dose-intensive chemotherapy with etoposide, carboplatin, cyclophosphamide and doxorubicin, administered for 34 weeks.
ResultsTables 3 and 4 present the characteristics of our patients. Their age ranged between 4 months and 15 years, with a median of 2.5 years. Twenty-three patients (57.5%) were male. The most frequent presentation was the presence of an abdominal mass (21 patients), followed by abdominal pain and chance finding (6 patients each), haematuria and fever (3 patients each) and lastly, in a patient with a syndrome associated with a high risk of cancer—WAGR syndrome—the tumour was found during an early cancer screen (1 patient).
Characteristics of patients at the time of diagnosis.
| PT | Age | Presentation | Biopsy | Anomalies | Post-CHEMO stage | Metastases | Post-CHEMO histology | Preoperative CHEMO | Surgery | Postoperative CHEMO |
|---|---|---|---|---|---|---|---|---|---|---|
| 1. M | 5 years | Abdominal mass | Yes | No | I | No | Regressive | Yes | Nephrectomy | – |
| 2. F | 2 years | Abdominal mass | No | WAGR | I | No | Stromal | Yes | Nephrectomy | AV1 |
| 3. M | 2 years | Abdominal pain | No | No | I | No | Blastemal | Yes | Nephrectomy | AVD |
| 4. M | 3 years | Abdominal pain+fever | No | No | II | No | Stromal | Yes | Nephrectomy | AV-2 |
| 5. M | 2 years | Abdominal pain+fever | No | No | I | No | Mixed | Yes | Nephrectomy | AV1 |
| 6. M | 1 year | Abdominal mass | No | No | I | No | Stromal | Yes | Nephrectomy | AV1 |
| 7. F | 1 year | Abdominal mass+fever | No | No | I | No | Stromal | Yes | Nephrectomy | AV1 |
| 8. M | 15 years | Abdominal mass | No | No | I | Pulmonary, only on CT scan | Diffuse anaplasia | No | Nephrectomy | AVD |
| 9. M | 5 years | Asthenia+weight loss+Abdominal mass | Yes | No | IV | Pulmonary+pleural+peritoneal | Regressive | Yes | Nephrectomy | AVD |
| 10. M | 3 years | Chance finding | No | No | V | Pulmonary+contralateral nephrogenic rests | Mixed | Yes | Right nephrectomy+left partial resection. | AVD |
| 11. F | 4 years | Abdominal mass | No | No | II | Pulmonary only on CT scan | Mixed | Yes | Nephrectomy | AV-2 |
| 12. F | 2 years | Abdominal mass+irritability | No | No | III | No | Stromal | Yes | Nephrectomy | AV-2 |
| 13. F | 1 year | Screening | No | WAGR | III | No | Mixed | Yes | Nephrectomy | AV -2 |
| 14. F | 3 years | Haematuria | No | No | I | No | Regressive | Yes | Nephrectomy | AV1 |
| 15. M | 9 months | Fever+irritability | No | Hemihypertrophy | I | No | Regressive | Yes | Nephrectomy | AV1 |
| 16. F | 2 years | Chance finding | No | Ataxia-telangiectasia | III | No | Blastemal | No | Nephrectomy | VP+CARBO +CYCLO+DOXO |
| 17. F | 3 years | Haematuria | No | No | III | No | Blastemal | Yes | Nephrectomy | VP+CARBO+CYCLO+DOXO |
| 18. F | 4 years | Chance finding | No | No | III | No | Regressive | Yes | Nephrectomy | AVD |
| 19. F | 3 years | Abdominal mass | No | No | III | No | Stromal | Yes | Nephrectomy | AVD |
| 20. M | 5 years | Abdominal mass | No | No | III | No | Regressive | Yes | Nephrectomy | AV-2 |
| 21. F | 3 years | Abdominal mass | No | No | II | No | Mixed | Yes | Nephrectomy | AVD |
| 22. F | 1 year | Abdominal mass | No | No | II | No | Clear cell sarcoma | Yes | Nephrectomy | VP+CARBO+CYCLO+DOXO |
| 23. M | 4 years | Abdominal mass | No | No | I | No | Blastemal | Yes | Nephrectomy | AVD |
| 24. F | 6 years | Fever | No | No | I | No | Mixed | Yes | Nephrectomy | AV1 |
| 25. M | 9 months | Abdominal mass | No | No | I | No | Epithelial | Yes | Nephrectomy | AV1 |
| 26. F | 15 years | Fever | No | No | II | No | Blastemal with diffuse anaplasia | No | Nephrectomy | AVD |
| 27. M | 4 months | Haematuria | No | No | I | No | Mixed | Yes | Nephrectomy | AV1 |
| 28. F | 7 years | Abdominal mass | No | No | I | No | Regressive | Yes | Nephrectomy | AV1 |
| 29. F | 5 years | Abdominal pain | No | No | I | No | Regressive | Yes | Nephrectomy | AV1 |
| 30. M | 7 years | Abdominal pain | No | No | III | Pulmonary | Necrotic | Yes | Nephrectomy | AV-2 |
| 31. M | 11 months | Abdominal pain | Yes | No | II | No | Mixed | Yes | Nephrectomy | AV2 |
| 32. F | 2 years | Abdominal mass | No | No | II | No | Mixed | Yes | Nephrectomy | AVD |
| 33. M | 3 years | Incidental | No | No | II | No | Regressive | Yes | Nephrectomy | AV1 |
| 34. M | 2 years | Abdominal mass+fever | No | No | II | No | Mixed | Yes | Nephrectomy | AVD |
| 35. M | 4 years | Chance finding | No | No | IV | Pulmonary | Blastemal | Yes | Nephrectomy | VP+CARBO+CYCLO+DOXO |
| 36. M | 1 year | Abdominal mass | No | No | V | NO | Blastemal | Yes | Right nephrectomy+left partial resection | VP+CARBO+CYCLO+ DOXO |
| 37. M | 2 years | Chance finding | No | No | I | No | Cystic | Yes | Nephrectomy | – |
| 38. F | 1 year | Abdominal mass | No | No | III | No | Stromal | Yes | Nephrectomy | AVD |
| 39. M | 1 year | Abdominal mass | No | No | I | No | Necrotic | Yes | Nephrectomy | – |
| 40. M | 2 years | Abdominal mass | No | No | I | No | Mixed | Yes | Nephrectomy | AV1 |
| PT | Radiotherapy | Status | Duration of followup | Relapse |
|---|---|---|---|---|
| 1. M | – | Alive | – | No |
| 2. F | No | Alive | 15 months | No |
| 3. M | No | Alive | 13 months | No |
| 4. M | No | Alive | 10 months | No |
| 5. M | No | Alive | 28 months | No |
| 6. M | No | Alive | 36 months | No |
| 7. F | No | Alive | 37 months | No |
| 8. M | No | Alive | 41 months | No |
| 9. M | Abdominal | Alive | 48 months | No |
| 10. M | Flank+contralateral kidney | Alive | 48 months | Lung |
| 11. F | No | Alive | 49 months | Thrombus in vena cava |
| 12. F | Flank | Alive | 52 months | No |
| 13. F | Flank | Alive | 64 months | No |
| 14. F | No | Alive | 67 months | No |
| 15. M | No | Alive | 76 months | No |
| 16. F | No | Deceased | 11 months | Local+metastatic (liver+lung) |
| 17. F | Flank | Alive | 81 months | No |
| 18. F | Flank | Alive | 85 months | No |
| 19. F | Flank | Alive | 87 months | No |
| 20. M | Flank | Alive | 81 months | No |
| 21. F | No | Alive | 81 months | No |
| 22. F | Flank | Alive | 89 months | No |
| 23. M | No | Alive | 97 months | No |
| 24. F | No | Alive | 101 months | No |
| 25. M | No | Alive | 106 months | No |
| 26. F | No | Alive | 112 months | No |
| 27. M | No | Alive | 112 months | No |
| 28. F | No | Alive | 120 months | No |
| 29. F | No | Deceased | 28 months | Local |
| 30. M | No | Alive | 120 months | No |
| 31. M | No | Alive | 127 months | No |
| 32. F | No | Alive | 128 months | No |
| 33. M | No | Alive | 130 months | No |
| 34. M | No | Lost to followup | 36 months | No |
| 35. M | No | Alive | 144 months | No |
| 36. M | No | Lost to followup | 46 months | No |
| 37. M | No | Alive | 148 months | No |
| 38. F | Flank | Alive | 148 months | No |
| 39. M | No | Alive | 154 months | No |
| 40. M | No | Alive | 167 months | No |
AV1, chemotherapy regimen including actinomycin D+vincristine; AVD: actinomycin D+vincristine+doxorubicin; CARBO, carboplatin; CHEMO, chemotherapy; CYCLO, cyclophosphamide; DOXO, doxorubicin; F, female; M, male; PT, patient; VP: etoposide.
Characteristics of patients with tumour recurrence.
| Age Sex | Post-chemo staging | Post-chemo histology | Surgery | Post-surgical Tx | Timea and site of recurrence | Second-line Tx | Status | Months since relapse |
|---|---|---|---|---|---|---|---|---|
| 3 years M | IV Lung metastases and nephrogenic rests in contralateral kidney | Mixed | Right nephrectomy+partial left nephrectomy | AVD+radiotherapy | 17 months Lung | Chemo+Rt+autologous HSCT | Alive | 31 months |
| 4 years F | II Lung metastases (only on CT scan) | Mixed | Nephrectomy | AV-2 | 13 months Thrombus in vena cava | Surgery+chemo+Rt+autologous HSCT | Alive | 26 months |
| 2 years F | III | Blastemal | Nephrectomy | VP+CARBO+CYCLO+DOXO | 9 months Local and metastatic (liver and lung) | Topotecan+ICE | Deceased | Disease progression: death at 11 months from initial diagnosis |
| 5 years F | I | Regressive | Nephrectomy | AV1 | 7 months Local | Chemo+Rt+autologous HSCT | Deceased | 2 subsequent recurrences: death 28 months after diagnosis |
| 9 years F | III | Regressive | Nephrectomy | AVD+radiotherapy | 15 months Abdominal | Surgery+Chemo++Rt+autologous HSCT | Alive | 60 months |
| 4 years M | I | Epithelial | Nephrectomy | AV1 | 15 months Lung | Surgery +Chemo+Rt+autologous HSCT | Alive | 58 months |
AV1, chemotherapy regimen including actinomycin D+vincristine; AVD: actinomycin D+vincristine+doxorubicin; CARBO, carboplatin; CYCLO, cyclophosphamide; DOXO, doxorubicin; chemo, chemotherapy; F, female; HSCT, haematopoietic stem cell transplantation; ICE, ifosfamide+etoposide+carboplatin; M, male; Rt: radiation therapy; Tx, treatment; VP, etoposide.
In our series, 4 patients had congenital anomalies: 2 had WAGR syndrome, 1 isolated hemihypertrophy and 1 ataxia-telangiectasia. Three of them responded well to treatment and are currently in first complete remission. The patient with ataxia-telangiectasia had both local and metastatic recurrence (lung and liver) refractory to chemotherapy, and died 2 months after relapsing. The poor outcome in this patient may have been associated to the need to reduce the chemotherapy dose by 25% and the contraindication of radiation therapy due to the underlying disease.
Only 3 patients required a biopsy due to their atypical clinical and radiological presentation: one aged less than 1 year at onset (11 months) and the other 2 presented with symptoms of urinary tract infection. None of the patients experienced complications from the biopsy.
Preoperative chemotherapy was given to 37 patients, who tolerated it well, and achieved a mean reduction in tumour volume of 250mL. Only 1 patient experienced toxicity (febrile neutropaenia) from chemotherapy.
In all patients, treatment of the primary tumour consisted in radical nephrectomy. In 3 patients, the operation was performed upfront, without prior chemotherapy. In a patient with bilateral Wilms tumour, the most affected kidney was completely removed, followed by partial resection of the second kidney at a later time. Another patient was originally classified as having a stage IV tumour with nephrogenic rests in the contralateral kidney, and later received a bilateral Wilms tumour diagnosis after surgery, where he underwent radical nephrectomy of one kidney with collection of a sample for biopsy from the contralateral kidney (patient no 10). This patient was treated as if he had a stage IV tumour, with 3-drug chemotherapy and radiation therapy in the affected flank and contralateral kidney.
The distribution by stages was the following: stage I, 18 patients (45%); stage II, 9 patients (22.5%); stage III, 9 patients (22.5%); stage IV, 2 patients (5%), and stage V, 2 patients (5%).
When it came to histology, based on the revised SIOP classification of renal tumours of childhood (2001), the most frequent histological subtypes corresponded to the intermediate risk group (11 mixed, 9 regressive, 7 stromal and 1 epithelial), with only 3 patients in the low-risk group (1 cystic, 2 necrotic) and 9 patients in the high-risk group (7 blastemal type, 1 diffuse anaplasia and 1 clear cell sarcoma).
Only 10 patients received radiation therapy: 8 exclusively to the flank of the affected kidney, one to the affected flank and the contralateral kidney, and 1 to the entire abdomen (on account of peritoneal dissemination). None of the patients received lung radiation therapy as first-line treatment.
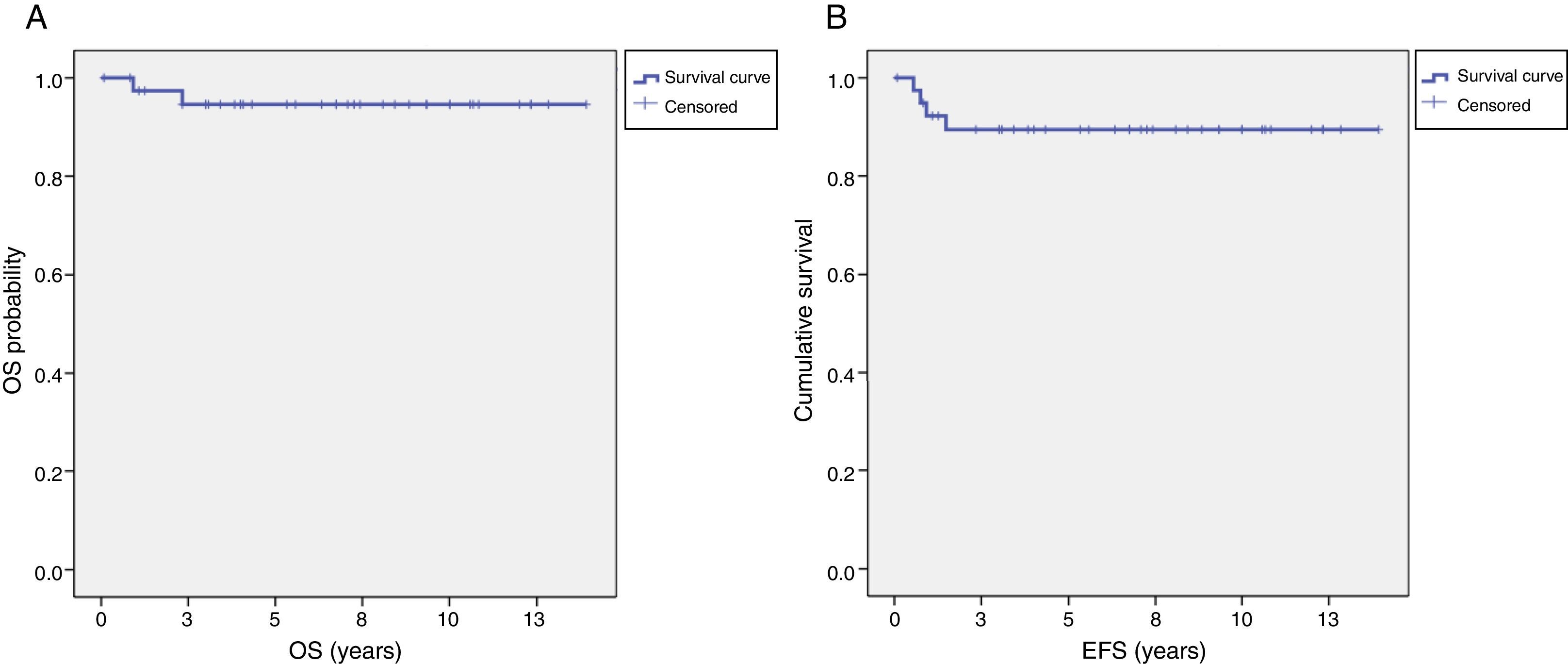
The median duration of followup was 6.75 years (range, 10 months–13.92 years). Two patients died due to disease progression. None died from chemotherapy toxicity or complications of surgery. The overall 5-year survival was 94.6±3.7% and the 5-year event-free survival was 89.4±5% (Fig. 1).
Fifteen percent of patients (6 out of 40) had disseminated disease at the time of diagnosis: 5 had pulmonary metastases and 1 had metastases in the lung, pleura and peritoneum. Two patients had recurrences (local and metastatic) and responded well to salvage therapy, remaining in second complete remission at the time of the last followup.
When it came to recurrence (Table 4), there were 6 patients in our series who had relapses; 4 of them were managed in our hospital from the time of diagnosis, while the other 2 were referred to the hospital when the tumour recurred. The median time elapsed between diagnosis and recurrence was 14 months (range, 7–17 months). Patients with recurrence were treated with chemotherapy, surgery, radiation therapy and autologous haematopoietic stem cell transplantation. Two patients died due to disease progression: one after the third recurrence and the other after the first recurrence, 28 and 11 months after the initial diagnosis, respectively. The first patient had blastemal histology since the initial diagnosis and suffered from ataxia-telangiectasia, which precluded the administration of the full treatment that corresponded to her risk group (as we noted above). The second patient had regressive histology at diagnosis and developed blastemal histology in the first recurrence. The other 4 patients remained alive at the time of the study, with a median duration of followup since the recurrence of 45 months (range, 26–60 months). Thus, in our case series, 66% of patients that had a recurrence remain in second complete remission.
DiscussionIn this article, we analyse our experience of the past 15 years in the management of Wilms tumour. The prognosis of nephroblastoma is very good, and the greatest challenge we face at present is to achieve a cure with minimum toxicity while preserving adequate renal function.6,17–19 In Europe, treatment consists of preoperative chemotherapy, followed by surgical excision with or without postoperative chemotherapy/radiation therapy depending on the stage and histology of the disease.6,20
Wilms tumour is diagnosed based on clinical and radiological features, and histological examination is usually not required to initiate treatment. In our series, only 3 patients required a biopsy before starting treatment, which confirmed the diagnosis of Wilms tumour in all.11,12
Preoperative chemotherapy usually achieves a reduction in tumour size that facilitates surgery without major toxicity. In our study, we found that preoperative treatment achieved a reduction in tumour size of 250mL in 90% of patients with little toxicity; 4 patients did not respond to this treatment.
Ongoing advances in our knowledge of the genetic and molecular basis of Wilms tumour will allow the future development of risk-adapted therapies better fitting individual patients, and to identify novel therapeutic targets with a more favourable efficacy/toxicity profile compared to standard chemotherapeutics.8 Some of the most important genetic changes described in the literature include the loss of heterozygosity for chromosomes 1p or 16q6 and the gain of chromosome 1q (which is the most frequent genetic change, found in up to 30% of patients). These changes are associated with less favourable outcomes and an increased risk of relapse and death.
Other mutations that lead to loss of function in tumour-suppressor and transcription factor genes involve the genes WT1, WT2, p53, FWT1, and FWT2, DROSHA or DICER1. Mutations that inactivate the WT1 gene or lead to loss of expression of the WT2 gene may lead to the persistence of nephrogenic rests that, for reasons yet unknown, can transform into Wilms tumour.8,10,13
While there have been significant advances in patient stratification, the presence of anaplastic histology continues to be the most important adverse prognostic feature.8
In our series, the overall 5-year survival in patients with localised as well as disseminated disease was excellent, consistent with similar studies published to date.3,4,17,19,20
Most recurrences of Wilms tumour occur within 2 years of diagnosis.21 The most frequent sites of recurrence are the lungs, abdomen and liver. Late recurrence (>5 years after diagnosis) is infrequent, occurring in 0.5% of patients, and has a similar prognosis to early recurrence.22 Recurrence survival is stratified based on risk factors (tumour histology and treatment received), and is reported in the literature as 34–64% (range, 15–85%),8,10,21,23 similar to the 66% observed in our series (4 of 6 patients). The factors that have a negative impact on survival are: greater age at diagnosis, advanced stage of disease and unfavourable histology. When it came to age at diagnosis, the 2 patients who died in our series had been 2 and 5 years old. As for advanced stage of disease, all patients that had metastatic disease at diagnosis were in complete remission at the end of the study period. Lastly, as regards histology, of the 2 patients who died, 1 had blastemal histology (the patient with ataxia-telangiectasia) and 1 regressive histology.
Patients with Wilms tumour require life-long followup, because most of them end up having a single kidney, and there is risk of developing late effects of treatment or second tumours (with a risk of a second malignant neoplasm of 5–7% at 30 years).4,6,22,24–26 Renal function must be monitored in patients with Wilms tumour on account of having a sole functioning kidney, which puts them at risk of renal failure due to the potential nephrotoxicity of chemotherapy and glomerular hyperfiltration in the nephrons left after surgery.2,8,26 The probability of experiencing terminal renal failure within 20 years of diagnosis is of 0.6% in patients in the early stages of disease, increases in those with bilateral disease or associated genetic syndromes, and reaches up to 74% in patients with Denys-Drash syndrome.5,24
To conclude, despite the considerable advances made in the diagnosis and treatment of these tumours in the past few decades and the high rate of survival, we cannot forget that patients continue to experience undesirable toxicity. In this sense, research efforts should aim towards minimising the side effects of treatment and to investigate new therapeutic targets, such as IGF-1 receptor inhibitors, antiangiogenic compounds, mTOR, JAK2 or telomerase inhibitors that could reduce the overall toxicity of treatment.1
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Illade L, Hernandez-Marques C, Cormenzana M, Lassaletta A, Andión Catalán M, Ruano D, et al. Tumor de Wilms: revisión de nuestra experiencia en los últimos 15 años. An Pediatr (Barc). 2018;88:140–149.
