







El tumor de Wilms es el tumor renal más frecuente en la edad pediátrica. Su tratamiento es multimodal: incluye quimioterapia y cirugía, con o sin radioterapia. La supervivencia de estos pacientes es excelente, superando el 90%. Presentamos la experiencia de nuestro centro en el tratamiento del tumor de Wilms durante los últimos 15 años.
Pacientes y métodosSe ha realizado un estudio retrospectivo de 40 pacientes pediátricos diagnosticados de forma consecutiva de nefroblastoma entre 2002 y 2016 en el Servicio de Hemato-Oncología pediátrica del Hospital Niño Jesús de Madrid. Se analizaron las características clínicas, los métodos diagnósticos, el tratamiento realizado y la evolución posterior.
ResultadosDe los 40 pacientes, 23 eran niños con una mediana de edad al diagnóstico de 2,5 años (rango, 4 meses-15 años). A 3 pacientes se les realizó nefrectomía inicial, 3 recibieron una punción aspiración con agua fina, seguida de quimioterapia y 34 pacientes recibieron quimioterapia preoperatoria directamente. La mediana de seguimiento de los pacientes fue de 6,75 años (rango, 10 meses-13,92 años).
Dos pacientes fallecieron de progresión de su enfermedad. Ningún paciente falleció de toxicidad en relación con el tratamiento. La supervivencia global y la supervivencia libre de evento a los 5 años fue del 94,6±3,7% y 89,4±5%, respectivamente.
ConclusiónEl tratamiento del tumor de Wilms es un éxito de la medicina moderna, consiguiendo en la actualidad una supervivencia que en nuestra serie alcanza el 95%.
Wilms’ tumour is the most frequent renal tumour in children. Multi-modal treatment includes chemotherapy and surgery, with or without radiotherapy. The survival is excellent, with rates exceeding 90%. A review is presented on our experience over the last 15 years of treating Wilms’ tumour in Hospital Niño Jesús, Madrid.
Patients and methodsA retrospective study was conducted on 40 consecutive paediatric patients diagnosed with nephroblastoma between 2002 and 2016 in the Hospital Niño Jesús in Madrid. The clinical characteristics, diagnostic methods, treatment, and follow-up were analysed.
ResultsOf the 40 patients, 23 were boys, with a median age at diagnosis of 2.5 years (range, 4 months-15 years). Three patients underwent initial nephrectomy, three received a fine needle aspiration biopsy, followed by chemotherapy, and 34 patients started pre-operative chemotherapy directly. The median follow-up of the patients was 6.75 years (range, 10 months - 13.92 years).
Two patients died from disease progression. There were no treatment-related deaths. Overall survival and event-free survival at 5 years was 94.6±3.7% and 89.4±5%, respectively.
ConclusionWilms’ tumour treatment is a success of modern medicine, currently achieving a survival rate of 95% in our series.
El tumor de Wilms o nefroblastoma es el segundo cáncer intraabdominal más común de la infancia y la quinta neoplasia maligna más frecuente en la edad pediátrica. Representa aproximadamente el 6% de todos los cánceres pediátricos y es el tumor renal más frecuente (más del 95% del total de tumores renales pediátricos)1,2,3. El diagnóstico diferencial del tumor de Wilms incluye el neuroblastoma y otras neoplasias renales pediátricas, como el sarcoma de células claras, tumor rabdoide maligno y el carcinoma de células renales, cuyo pronóstico es menos favorable1,4.
Los pacientes con diversas anomalías congénitas, como el síndrome de WAGR, el síndrome de Denys-Drash, el síndrome de Beckwith-Wiedemann o la hemihipertrofia de forma aislada, tienen un mayor riesgo de desarrollar tumor de Wilms3.
El pico de incidencia del nefroblastoma se produce entre los 2 y 5 años de edad, siendo el 95% de los niños diagnosticados antes de los 10 años5. El signo más frecuente es el hallazgo incidental de una masa abdominal, seguido de la presencia de hematuria. En algunos casos, puede asociar hipertensión arterial por isquemia renal secundaria a la presión ejercida sobre la arteria renal. En ocasiones, estos pacientes pueden presentar clínica constitucional con astenia, anorexia, pérdida de peso y fiebre3. Entre el 10 y el 25% de los pacientes pueden presentar diseminación a distancia, que suele ser pulmonar6.
El tratamiento de los tumores renales ha experimentado un avance espectacular desde la introducción del tratamiento multimodal, que incluye quimioterapia y cirugía con o sin radioterapia. Con una tasa de supervivencia a los 5 años que ha aumentado dramáticamente en las últimas 3 décadas, del 25% en la era prequimioterapia de finales de 1960 y principios de 1970, al 90% en la década de 19903,4,7.
Dos grandes grupos cooperativos se han involucrado en el desarrollo e investigación del tumor de Wilms: el National Wilms Tumor Study Group (NWTSG) en Estados Unidos y la Sociedad Internacional de Oncología Pediátrica (SIOP) en Europa. Ambos difieren en el momento en que realizan la cirugía. El NWTSG defiende la nefrectomía inmediata para asegurar el diagnóstico histológico y la estadificación precisa8-12. El Grupo de Estudio de Tumores Renales de la SIOP defiende la quimioterapia preoperatoria para promover la reducción del tumor y de este modo facilitar la cirugía —evitando rupturas de la cápsula tumoral—, realizando la estadificación tras la misma1,3,8. Ambos enfoques producen tasas de supervivencia global similares11,13,14. La realización de biopsia confirmatoria prequimioterapia en los casos de tumor de Wilms con características clínicas y radiológicas típicas no forma parte de la práctica habitual de los protocolos de la SIOP, ya que se han realizado estudios en los que se comparaban muestras de biopsia prequimioterapia con la nefrectomía realizada posteriormente a estos mismos pacientes, obteniéndose un 99,5% de concordancia13.
Dada la alta tasa de supervivencia alcanzada, el objetivo principal en el momento actual es individualizar el tratamiento de acuerdo con una correcta estratificación según los grupos de riesgo, con el fin de alcanzar los más altos índices de curación, disminuyendo la frecuencia y la intensidad de la toxicidad aguda y tardía derivada del tratamiento5.
Sobre la base de la correlación entre las características histológicas y la supervivencia, la SIOP ha definido 3 grupos pronósticos: bajo riesgo, riesgo intermedio y tumores de alto riesgo5,15. El marcador pronóstico desfavorable más importante es la histología anaplásica. Otras histologías incluidas en el grupo de alto riesgo son la blastematosa, el sarcoma de células claras y el tumor rabdoide renal10,13. El segundo factor más importante es el estadio del tumor. Como en la mayoría de los tumores, el estadio más bajo presenta mejor pronóstico.
Otros factores que contribuyen a la estratificación del riesgo del tumor de Wilms son la edad del paciente, el tamaño del tumor y la respuesta al tratamiento16. La determinación de estos factores incluirá al paciente en uno de los 3 riesgos y de esa forma será tratado2,7.
La supervivencia de los pacientes con tumor de Wilms es generalmente excelente, superando el 90%2. Para los pacientes en estadio i y ii es del 95%, en estadio iii es del 75-80% y en estadio iv un 65-75% (tabla 1). Solo el 15% de los pacientes con histología favorable tienen enfermedad recurrente, en comparación con una tasa del 50% en aquellos con histología anaplásica. Incluso los pacientes con enfermedad en recaída tiene una tasa de supervivencia alta, entorno al 60%7. Los sitios más comunes de recaída son los pulmones, pleura, lecho del tumor y el hígado. Entre los pacientes metastásicos, aquellos con afectación hepática tienen un peor pronóstico que aquellos con afectación pulmonar5.
Estadificación de la SIOP de los tumores renales de la infancia (2001)
| Estadio I | El tumor está limitado al riñón |
| No hay células tumorales en el margen quirúrgico | |
| Los vasos del seno renal no están implicados | |
| Los vasos intrarrenales pueden estar involucrados la cápsula, los tejidos adyacentes, vasos del seno renal y vena cava | |
| Estadio II | El tumor se extiende fuera del riñón pero es totalmente resecado: la cápsula, los tejidos adyacentes, vasos del seno renal y vena cava pueden estar implicados |
| Estadio III | Resección incompleta: |
| – afectación de ganglios linfáticos intraabdominal | |
| – rotura del tumor antes o durante la cirugía | |
| – afectación del peritoneo/implantes peritoneales | |
| – trombo tumoral presente en los márgenes de resección | |
| Diseminación hematógena (pulmón, hígado, hueso…) | |
| Estadio IV | Afectación de ganglios extra-abdominales |
| Estadio V | Tumor renal bilateral (cada riñón debe ser estadificado por separado) |
Durante un periodo de 15 años (1 de enero del 2002 a 1 junio del 2016), 40 pacientes fueron diagnosticados y tratados de nefroblastoma en el Servicio de Hemato-Oncología pediátrica del Hospital Infantil Universitario Niño Jesús. A esta serie añadimos 2 pacientes más, que fueron derivados a nuestro centro tras recaer de su enfermedad y que se incluyen en el análisis de los pacientes con recaídas.
Se revisaron las historias clínicas de los pacientes para obtener información sobre edad, sexo, clínica al diagnóstico, histología, estadio tumoral, supervivencia global y supervivencia libre de evento. Se evaluaron además, el tratamiento recibido, el tipo de recaída y el tratamiento de la recaída.
Las investigaciones incluyeron análisis de sangre, determinación de la creatinina y electrolitos en sangre y la función hepática. Estos test se realizaron antes de comenzar y durante el tratamiento quimioterápico. Se completó el estudio con ecografía abdominal y radiografía o TAC pulmonar. Se realizó RM en la mayoría de los casos o TAC abdominal para evaluar el tamaño tumoral, el estadio y la operabilidad.
En todos los pacientes se realizó clasificación histológica y estadificación según el protocolo de la SIOP (tablas 1 y 2).
Clasificación revisada de la SIOP de los tumores renales de la infancia (2001)
| Tumores de bajo riesgo |
| Nefroblastoma quístico parcialmente diferenciado |
| Nefroblastoma completamente necrosado |
| Tumores de riesgo intermedio |
| Nefroblastoma tipo epitelial |
| Nefroblastoma tipo estroma |
| Nefroblastoma tipo mixto |
| Nefroblastoma tipo regresivo |
| Nefroblastoma con anaplasia focal |
| Tumores de alto riesgo |
| Nefroblastoma tipo blastematoso |
| Nefroblastoma con anaplasia difusa |
| Sarcoma de células claras del riñón |
| Tumor rabdoide del riñón |
Todos los pacientes recibieron tratamiento según el protocolo para nefroblastoma de la SIOP 2001. De acuerdo con el mismo, los pacientes recibieron quimioterapia preoperatoria seguida de cirugía, excepto 3 pacientes en los que se realizó nefrectomía directamente al diagnóstico. En 3 ocasiones se realizó punción-aspiración con aguja fina de la lesión previo a iniciar tratamiento (ya que se desaconseja la biopsia excisional pues puede aumentar la estatificación tumoral), por su presentación clínica o radiológica inusual. En todos ellos se obtuvo confirmación histológica de nefroblastoma tras la cirugía.
Los pacientes con enfermedad localizada recibieron 4 semanas de quimioterapia estándar con actinomicina D y vincristina. Los pacientes con enfermedad avanzada recibieron 6 semanas de un régimen de 3 fármacos con actinomicina D, vincristina y doxorrubicina. Tras la quimioterapia inicial, se realizaron una TAC o RM para evaluar la respuesta tumoral y programar la cirugía. La respuesta tumoral se valoró en relación con la disminución del diámetro tumoral máximo entre las imágenes pre y posquimioterapia.
En relación con el tratamiento poscirugía, 3 pacientes pertenecían al estadio i de bajo riesgo, por lo que no precisaron más tratamiento. El resto recibió quimioterapia poscirugía entre 4 y 34 semanas. El régimen de quimioterapia se basó en el estadio tumoral y la histología.
Los pacientes con enfermedad limitada al órgano con histología favorable recibieron actinomicina D y vincristina durante 4 semanas. Los pacientes con enfermedad más avanzada recibieron actinocimina D, y vincristina con o sin doxorrubicina durante 27 semanas. En los pacientes con hallazgos histológicos desfavorables, la quimioterapia intensiva consistió en etopósido, carboplatino, ciclofosfamida y doxorrubicina, durante 34 semanas.
ResultadosEn las tablas 3 y 4 se muestran las características de los pacientes. El rango de edad de los pacientes varía desde los 4 meses hasta los 15 años, con una mediana de 2,5 años. De ellos, 23 pacientes (57,5%) eran varones. El síntoma de presentación más frecuente fue la presencia de una masa abdominal (21 pacientes), seguido de dolor abdominal y hallazgo incidental (ambos 6 pacientes), hematuria y fiebre (ambos 3 pacientes) y, por último, en un paciente con síndrome de predisposición al cáncer —síndrome de WARG— durante un screening para la detección del cáncer precoz (un paciente).
Características de los pacientes al diagnóstico
| Pac | Edad | Clínica | Biopspia | Anomalías | Estadio post-Qt | Metástasis | Histología post-Qt | QT pre | Cirugía | QT post |
|---|---|---|---|---|---|---|---|---|---|---|
| 1. M | 5 años | Masa abdominal | Sí | No | I | No | Regresivo | Sí | Nefrectomía | – |
| 2. F | 2 años | Masa abdominal | No | Warg | I | No | Estromal | Sí | Nefrectomía | AV1 |
| 3. M | 2 años | Dolor abdominal | No | No | I | No | Blastematoso | Sí | Nefrectomía | AVD |
| 4. M | 3 años | Dolor abdominal+fiebre | No | No | II | No | Estromal | Sí | Nefrectomía | AV-2 |
| 5. M | 2 años | Dolor abdominal+fiebre | No | No | I | No | Mixto | Sí | Nefrectomía | AV 1 |
| 6. M | 1 años | Masa abdominal | No | No | I | No | Estromal | Sí | Nefrectomía | AV1 |
| 7. F | 1 años | Masa abdominal+fiebre | No | No | I | No | Estromal | Sí | Nefrectomía | AV1 |
| 8. M | 15 años | Masa abdominal | No | No | I | Pulmonares solo en TC | Anaplasia difusa | No | nefrectomía | AVD |
| 9. M | 5 años | Astenia+pérdida de peso+masa abdominal | Sí | No | IV | Pulmonares+pleurales + peritoneales | Regresivo | SI | nefrectomía | AVD |
| 10. M | 3 años | Incidental | No | No | V | Pulmonares+restos nefrogénicos en contralateral | Mixto | Sí | Nefrectomía dcha + resección focal izq. | AVD |
| 11. F | 4 años | Masa abdominal | No | No | II | pulmonares sólo en TC | Mixto | Sí | Nefrectomía | AV-2 |
| 12. F | 2 años | Masa abdominal + irritabilidad | No | No | III | NO | Estromal | Sí | Nefrectomía | AV-2 |
| 13. F | 1 años | Screening | No | Warg | III | No | Mixto | Sí | Nefrectomía | AV -2 |
| 14. F | 3 años | Hematuria | No | No | I | No | Regresivo | Sí | Nefrectomía | AV1 |
| 15. M | 9 meses | Fiebre+irritabilidad | No | Hemihipertrofia | I | No | Regresivo | Sí | Nefrectomía | AV1 |
| 16. F | 2 años | Incidental | No | Ataxia-telangiectasia | III | No | Blastematoso | No | Nefrectomía | VP+CARBO +CICLO+DOXO |
| 17. F | 3 años | Hematuria | No | No | III | No | Blastematoso | Sí | Nefrectomía | VP+CARBO + CICLO+DOXO |
| 18. F | 4 años | Incidental | No | No | III | No | Regresivo | Sí | Nefrectomía | AVD |
| 19. F | 3 años | Masa abdominal | No | No | III | No | Estromal | Sí | Nefrectomía | AVD |
| 20. M | 5 años | Masa abdominal | No | No | III | No | Regresivo | Sí | Nefrectomía | AV-2 |
| 21. F | 3 años | Masa abdominal | No | No | II | No | Mixto | Sí | Nefrectomía | AVD |
| 22. F | 1 años | Masa abdominal | No | No | II | No | Sarcoma de células claras | Sí | Nefrectomía | VP + CARBO + CICLO + DOXO |
| 23. M | 4 años | Masa abdominal | No | No | I | No | Blastematoso | Sí | Nefrectomía | AVD |
| 24. F | 6 años | Fiebre | No | No | I | No | Mixto | Sí | Nefrectomía | AV1 |
| 25. M | 9 meses | Masa abdominal | No | No | I | No | Epitelial | SI | nefrectomía | AV1 |
| 26. F | 15 años | Fiebre | No | No | II | No | Blastematoso con anaplasia difusa | NO | nefrectomía | AVD |
| 27. M | 4 meses | Hematuria | No | No | I | No | Mixto | Sí | Nefrectomía | AV1 |
| 28. F | 7 años | Masa abdominal | No | No | I | No | Regresivo | Sí | Nefrectomía | AV1 |
| 29. F | 5 años | Dolor abdominal | No | No | I | No | Regresivo | Sí | Nefrectomía | AV1 |
| 30. M | 7 años | Dolor abdominal | No | No | III | Pulmonares | Necrosado | Sí | Nefrectomía | AV-2 |
| 31. M | 11 meses | Dolor abdominal | Sí | No | II | No | Mixto | Sí | Nefrectomía | AV2 |
| 32. F | 2 años | Masa abdominal | No | No | II | No | Mixto | Sí | Nefrectomía | AVD |
| 33. M | 3 años | Incidental | No | No | II | No | Regresivo | SI | Nefrectomía | AV1 |
| 34. M | 2 años | Masa abdominal+fiebre | No | No | II | No | mixto | SI | Nefrectomía | AVD |
| 35. M | 4 años | Incidental | No | No | IV | Pulmonares | Blastematoso | Sí | Nefrectomía | VP+CARBO + CICLO+DOXO |
| 36. M | 1 años | Masa abdominal | No | No | V | NO | Blastematoso | Sí | Nefrectomía dcha + resección focal izq | VP + CARBO + CICLO+ DOXO |
| 37. M | 2 años | Incidental | No | No | I | No | Quístico | Sí | Nefrectomía | – |
| 38. F | 1 años | Masa abdominal | No | No | III | No | Estromal | Sí | Nefrectomía | AVD |
| 39. M | 1 años | Masa abdominal | No | No | I | No | Necrosado | Sí | Nefrectomía | – |
| 40. M | 2 años | Masa abdominal | No | No | I | No | Mixto | Sí | Nefrectomía | AV1 |
| Pac | Radioterapia | Status | Tiempo de seguimiento | Recaída |
|---|---|---|---|---|
| 1. M | – | Vivo | – | No |
| 2. F | No | Viva | 15 meses | No |
| 3. M | No | Vivo | 13 meses | No |
| 4. M | No | Vivo | 10meses | No |
| 5. M | No | Vivo | 28 meses | No |
| 6. M | No | Vivo | 36 meses | No |
| 7. F | No | Viva | 37 meses | No |
| 8. M | No | Vivo | 41 meses | No |
| 9. M | Abdominal | Vivo | 48 meses | No |
| 10. M | Flanco+riñón contralateral | Vivo | 48 meses | Pulmón |
| 11. F | No | Viva | 49 meses | Trombosis vena cava |
| 12. F | Flanco | Viva | 52 meses | No |
| 13. F | Flanco | Viva | 64 meses | No |
| 14. F | No | Viva | 67 meses | No |
| 15. M | No | Vivo | 76 meses | No |
| 16. F | No | Defunción | 11 meses | Local+metástasis (hígado+pulmón) |
| 17. F | Flanco | Viva | 81 meses | No |
| 18. F | Flanco | Viva | 85 meses | No |
| 19. F | Flanco | Viva | 87 meses | No |
| 20. M | Flanco | Vivo | 81 meses | No |
| 21. F | No | Viva | 81 meses | No |
| 22. F | Flanco | Viva | 89 meses | No |
| 23. M | No | Vivo | 97 meses | No |
| 24. F | No | Viva | 101 meses | No |
| 25. M | No | Viva | 106 meses | No |
| 26. F | No | Viva | 112 meses | No |
| 27. M | No | Vivo | 112 meses | No |
| 28. F | No | Viva | 120 meses | No |
| 29. F | No | Defunción | 28 meses | Local |
| 30. M | No | Vivo | 120 meses | No |
| 31. M | No | Vivo | 127 meses | No |
| 32. F | No | Vivo | 128 meses | No |
| 33. M | No | Vivo | 130 meses | No |
| 34. M | No | Pérdida | 36 meses | No |
| 35. M | No | Viva | 144 meses | No |
| 36. M | No | Pérdida | 46 meses | No |
| 37. M | No | Vivo | 148 meses | No |
| 38. F | Flanco | Viva | 148 meses | No |
| 39. M | No | Vivo | 154 meses | No |
| 40. M | No | Vivo | 167 meses | No |
AV1: régimen de quimioterapia que incluye actinocimina D, vincristina; AVD: actinomicina D, vincristina, doxorrubicina; CARBO: carboplatino; CICLO: ciclofosfamida; dcha: derecha; DOXO: doxorrubicina; Estadio post-Qt: estadio posquimioterapia; F: femenino; izq: izquierdo; M: masculino; Pac: pacientes; Qt post: quimioterapia poscirugía; Qt pre: quimioterapia precirugía; VP: etopósido.
Características de los pacientes en reída
| Edad Sexo | Estadio post-Qt | Histología post-Qt | Cirugía | Tratamiento post-Cx | Tiempo y lugar recaídaa | T.° segunda línea | Status | Meses desde la recaída |
|---|---|---|---|---|---|---|---|---|
| 3años M | IV metástasis pulmonares y restos nefrogénicos riñón contralateral | Mixto | Nefrectomía derecha+resección focal izquierda | AVD + radioterapia | 17 meses Pulmón | Qt+Rt + TPH autógeno | Vivo | 31 meses |
| 4 años F | II metástasis pulmonares (solo en TAC) | Mixto | Nefrectomía | AV-2 | 13 meses Trombosis de la cava | Cirugía+Qt+Rt + TPH autógeno | Vivo | 26 meses |
| 2 años F | III | Blastematoso | Nefrectomía | VP+CARBO+CICLO+DOXO | 9 meses Local y metastásica (hígado y pulmón) | Topotecan + ICE | Defunción | Progresión de la enfermedad: muerte a los 11 meses del diagnóstico inicial |
| 5 años F | I | Regresivo | Nefrectomía | AV1 | 7 meses Local | Qt+Rt+TPH autógeno | Defunción | 2 recaídas posteriores: muerte 28 meses desde el diagnóstico |
| 9 años F | III | Regresivo | Nefrectomía | AVD+radioterapia | 15 meses Abdominal | Cirugía+ Qt+Rt + TPH autógeno | Vivo | 60 meses |
| 4 años M | I | Epitelial | Nefrectomía | AV1 | 15 meses Pulmón | Cirugía+Qt+Rt + TPH autógeno | Vivo | 58 meses |
AV1: régimen de quimioterapia que incluye actinomicina D, vincristina; AVD: actinomicina D, vincristina, doxorrubicina; CARBO: carboplatino; CICLO: ciclofosfamida; DOXO: doxorrubicina; Estadio post-Qt: estadio posquimiotrapia; F: femenino; ICE: ifosfamida + etopósido+carboplatino; M: masculino; Qt: quimioterapia; Rt: radioterapia; TPH autógeno: trasplante autógeno; Tratamiento post-Cx: tratamiento poscirugía; VP: etopósido.
En nuestra serie, 4 pacientes asociaban anomalías congénitas: 2 síndrome de WARG, uno hemihipertrofia aislada y otro ataxia-telangiectasia. Tres de ellos presentaron buena respuesta al tratamiento y se encuentran en primera remisión completa. La paciente con ataxia-telangiectasia presentó recaída local y metastásica (pulmonar y hepática), siendo refractario a la quimioterapia y falleciendo a los 2 meses de la recaída. La mala evolución de esta paciente podría estar asociada con la necesidad de reducción de dosis de la quimioterapia en un 25%, así como de evitar la administración de radioterapia, debido a su enfermedad de base.
Solo 3 pacientes precisaron biopsia por tener una presentación clínica- radiológica atípica: uno era menor de un año al comienzo (11 meses) y los otros 2 por presentar clínica de infección del tracto urinario. Ningún paciente presentó complicaciones relacionadas con la biopsia.
La quimioterapia preoperatoria fue administrada en 37 pacientes, siendo muy bien tolerada, con una reducción media del volumen tumoral de 250ml. Únicamente un paciente presentó toxicidad (neutropenia febril) con la quimioterapia.
El tratamiento local de todos los pacientes consistió en nefrectomía radical. En 3 pacientes esta se realizó directamente sin quimioterapia preoperatoria. En un caso de tumor de Wilms bilateral se realizó nefrectomía del riñón más afectado y resección parcial del otro riñón en un segundo tiempo. Otro paciente fue considerado inicialmente un estadio iv con restos nefrogénicos en el riñón contralateral, siendo diagnosticado de tumor de Wilms bilateral tras la cirugía, en la que se realizó nefrectomía de un riñón y toma de biopsia del riñón contralateral (paciente número 10). Este paciente recibió tratamiento como si se tratase de un estadio iv, con un régimen de poliquimioterapia con 3 fármacos, así como radioterapia en el flanco afectado y en el riñón contralateral.
La distribución por estadios fue la siguiente: estadio i, 18 pacientes (45%); estadio ii, 9 pacientes (22,5%); estadio iii, 9 pacientes (22,5%); estadio iv, 2 pacientes (5%), y estadio v, 2 pacientes (5%).
En relación con la histología, según la clasificación revisada de la SIOP de los tumores renales de la infancia (2001), el subtipo histológico más frecuente fue el de riesgo intermedio con 28 pacientes (11 mixtos, 9 regresivos, 7 estromales y uno epitelial), solo 3 pacientes eran de bajo riesgo (uno quístico, 2 necrosados) y 9 pacientes fueron de alto riesgo (7 blastematosos, una anaplasia difusa y una sarcoma de células claras).
Sólo 10 pacientes recibieron radioterapia: 8 recibieron radioterapia exclusivamente en el flanco del riñón afectado, uno recibió radioterapia en el flanco y en el riñón contralateral, y otro recibió radioterapia abdominal (por presentar diseminación peritoneal). Ningún paciente recibió radioterapia pulmonar en primera línea de tratamiento.
La mediana de seguimiento de los pacientes fue de 6,75 años (rango, 10 meses-13,92 años). Dos pacientes fallecieron por progresión de su enfermedad. Ningún paciente falleció de toxicidad en relación con la quimioterapia o la cirugía. La supervivencia global a los 5 años del fue 94,6±3,7% y la supervivencia libre de evento a los 5 años del 89,4±5% (fig. 1).
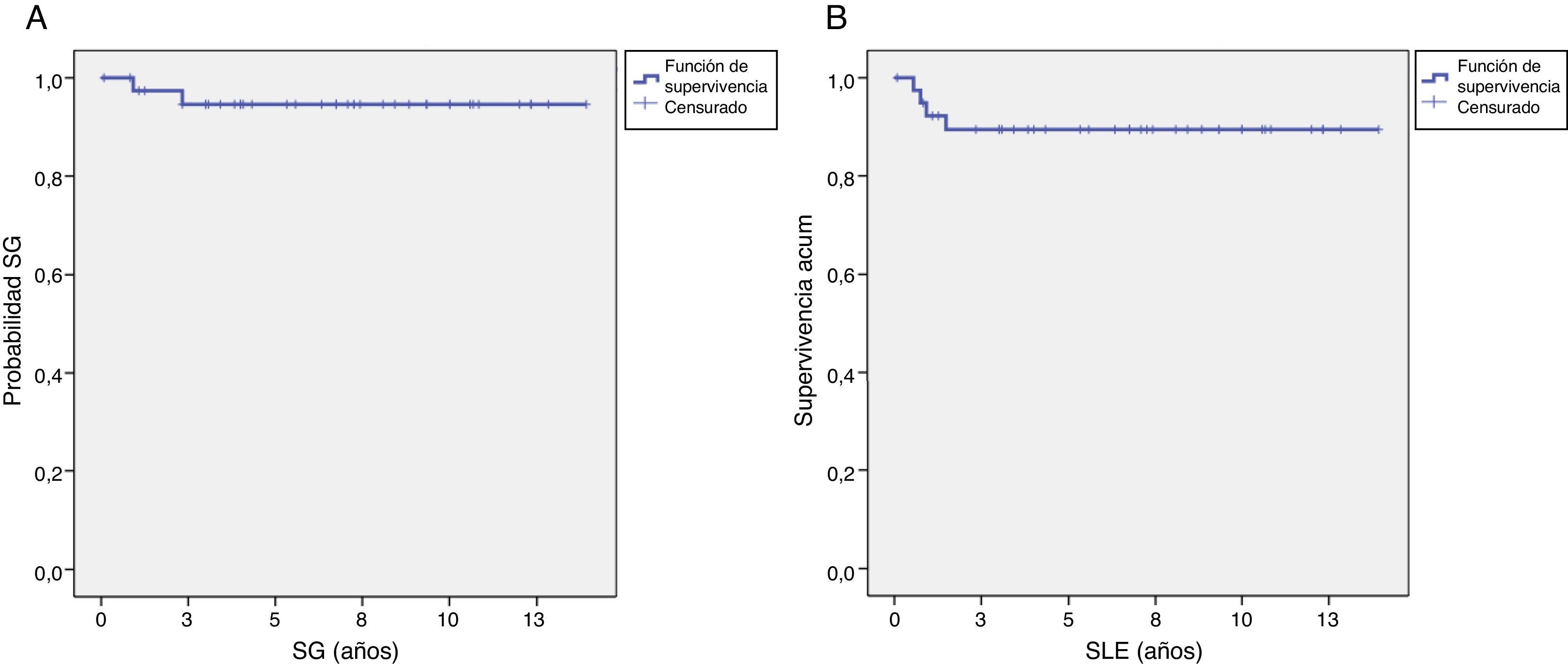
El 15% de los pacientes (6 de 40) presentaban enfermedad metastásica al diagnóstico, 5 en forma de metástasis pulmonares y uno que asociaba metástasis pulmonares, pleurales y peritoneales. Dos de ellos tuvieron recidiva de la enfermedad (local y metastásica), siendo ambos rescatados con tratamiento de segunda línea y permaneciendo en segunda remisión completa en el momento del último seguimiento.
En relación con las recaídas de la enfermedad (tabla 4), nuestra serie incluye 6 pacientes, 4 seguidos en nuestro centro desde el diagnóstico y 2 pacientes derivados de otros hospitales tras presentar recaída. La mediana de tiempo desde el diagnóstico hasta la recaída fue de 14 meses (rango 7-17 meses). Los pacientes en recaída recibieron tratamiento con quimioterapia, cirugía, radioterapia y trasplante autólogo. Dos pacientes fallecieron por progresión de su enfermedad: uno tras su 3.a y otro tras su 1-a recaída, a los 28 y 11 meses del diagnóstico inicial, respectivamente. El primero de ellos presentaba histología blastematosa desde el diagnóstico inicial, así como ataxia-telangiectasia, lo que impidió la administración del tratamiento completo que le correspondía según su grupo de riesgo (ya comentado en párrafos anteriores). El segundo paciente era de tipo regresivo al diagnóstico, desarrollando histología blastematosa en la primera recaída. Los otros 4 pacientes siguen vivos en el momento del estudio, con una mediana de seguimiento desde la recaída de 45 meses (rango 26-60 meses). Por lo tanto, en nuestra serie, el 66% de los pacientes que presentaron una recaída continúan en segunda remisión completa.
DiscusiónEn el presente artículo evaluamos nuestra experiencia en el manejo del tumor de Wilms durante los últimos 15 años. El nefroblastoma tiene muy buen pronóstico; el mayor desafío actual es alcanzar la curación con la menor toxicidad posible y preservar una adecuada función renal17-20. El tratamiento en Europa consiste en quimioterapia precirugía, seguido de extirpación quirúrgica con o sin quimioterapia/radioterapia postoperatoria en función del estadio y la histología de la enfermedad17,21.
El diagnóstico es clínico y radiológico, y la mayoría de las veces no es necesaria la comprobación histológica para iniciar el tratamiento. En nuestra serie, solo 3 pacientes precisaron biopsia previa a iniciar el tratamiento, y en todos ellos se confirmó la sospecha de tumor de Wilms11,12.
La quimioterapia preoperatoria suele conseguir una disminución del volumen tumoral que facilita la cirugía, sin toxicidades importantes. En nuestro estudio se observó poca toxicidad de la misma y una disminución media del tamaño tumoral de 250ml en el 90% de los pacientes; no se observó respuesta en 4 pacientes.
Los continuos avances en el conocimiento de las bases genéticas y moleculares del tumor de Wilms permitirán establecer en el futuro terapias «adaptadas al riesgo» individualizado de los pacientes, así como identificar nuevas dianas terapéuticas con un perfil de eficacia/toxicidad más favorable comparadas con la quimioterapia estándar8. Entre las alteraciones genéticas descritas más importantes se encuentran: la pérdida de heterocigosis de los cromosomas 1p o 16q 6, así como la ganancia del cromosoma 1q (que es la anormalidad genética más frecuente, presente hasta en el 30% de los pacientes). Estas anomalías se han asociado con un peor pronóstico, y un mayor riesgo de recaída y muerte.
Otras mutaciones que implican la pérdida de funcionalidad de una serie de genes supresores de tumores y de transcripción incluyen los genes WT1, WT2, p53, FWT1, y FWT2, DROSHA o DICER1. Las mutaciones que provocan la inactividad del gen WT1 o la pérdida de expresión del gen WT2 pueden provocar la persistencia de restos nefrogénicos que, por razones aún no conocidas, se transforman en tumor de Wilms 8,10,13.
Pese a todos estos avances en la clasificación de los pacientes, el factor de mal pronóstico más importante continúa siendo la presencia de histología anaplásica8.
En nuestra experiencia, la supervivencia global a 5 años, en pacientes con enfermedad tanto localizada como metastásica, fue excelente, en consonancia con otros estudios similares publicados hasta la fecha3,4,18,20,21.
La mayoría de las recaídas de tumor de Wilms ocurren en los 2 primeros años tras el diagnóstico22. Los sitios más frecuentes de recaída son el pulmón, el abdomen y el hígado. Las recaídas tardías de la enfermedad (5 años tras el diagnóstico) son poco frecuentes, pudiendo observarse en el 0,5% de los pacientes, con un pronóstico similar al de las recaídas precoces23. La supervivencia de las recaídas se estratifica en función de los factores de riesgo (histología tumoral y tratamiento recibido), de forma global en la literatura se sitúa en torno al 34-64% rango entre 15 y 85%8,10,22,24, similar a la observada en nuestra serie de un 66% (4 de 6 pacientes). Los factores que influyen negativamente en la supervivencia de los pacientes son: la mayor edad al diagnóstico, el estadio avanzado de la enfermedad y la histología desfavorable. En cuanto a la edad al diagnóstico, en nuestra serie los 2 pacientes que fallecieron tenían 2 y 5 años. En relación con el estadio avanzado de la enfermedad, todos los pacientes con enfermedad metastásica al diagnóstico se encuentran en remisión completa en la fecha de finalización del estudio; y por último, referido a la histología de la enfermedad, los 2 pacientes que fallecieron presentaban una histología de tipo blastematoso (el paciente con ataxia-telangiectasia) y el otro era de tipo regresivo.
Los pacientes con tumor de Wilms precisan seguimiento indefinido debido a su condición en la mayoría de los casos de monorrenos, por las posibles toxicidades de la quimioterapia y por la posibilidad de aparición de segundos tumores (con un riesgo de un 2o evento tumoral de entre 5-7% a los 30 años4,17,23,25-27. Es necesario monitorizar la función renal de los pacientes con tumor de Wilms debido a su condición de monorrenos, que les confiere riesgo de presentar insuficiencia renal, tanto por la nefrotoxicidad de la quimioterapia, como por el riesgo de hiperfiltración de las nefronas restantes tras la nefrectomía que se realiza para extirpar el tumor2,8,27. El riesgo de presentar insuficiencia renal terminal a los 20 años del diagnóstico es de un 0,6% en los pacientes con estadios más precoces de la enfermedad, va en aumento en aquellos que presentan enfermedad bilateral o síndromes genéticos asociados, alcanzando un 74% en los pacientes con síndrome de Denys-Drash5,25.
En conclusión, pese a los grandes avances diagnósticos y terapéuticos alcanzados en las últimas décadas en este tipo de tumores y la alta tasa de supervivencia, no hay que despreciar que los pacientes continúan presentando toxicidades no deseables. En este sentido los esfuerzos deben dirigirse a tratar de minimizar los efectos secundarios del tratamiento, así como a investigar nuevas dianas terapéuticas como los inhibidores del receptor de IGF1, compuestos antiangiogénicos, mTOR, JAK2 o inhibidores de la telomerasa, que pudieran disminuir la toxicidad global del tratamiento1.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
