
Autosomal recessive polycystic kidney disease (ARPKD), a rare inherited ciliopathy primarily involving the kidney and liver, is in most cases caused by pathogenic variants of the PKHD1 gene (locus 6p21.1-p12) encoding the fibrocystin protein.1 Historically, it has been considered a heterogeneous disease in terms of its presentation and outcomes, and a clear genotype-phenotype correlation has not been established.1
Focusing on the clinical presentation at onset and the course of disease during the paediatric age range, we reviewed the health records of 7 patients with a diagnosis of ARPKD, all with pathogenic variants of PKHD1, to describe the phenotype of the disease and attempt to contribute data on clinical aspects that have yet to be fully elucidated.
Genetic testing was carried out with a gene panel that included several genes contemplated in the differential diagnosis of ARPKD (PKD1, PKD2, PKHD1, UMOD, MUC1, REN, HNF1B, OFD1, TSC1, TSC2, COL4A3, COL4A4, COL4A5) with the Ion Torrent (IG S5 Sequencer, Thermo Fisher Scientific, Waltham, MA, USA) next generation sequencing (NGS) platform. Variants were identified with the Variant Caller version 5 software (Thermo Fisher Scientific, Waltham, MA, USA). All genetic findings were corroborated by means of Sanger sequencing.
The study was approved by the Research Ethics Committee of the Principality of Asturias.
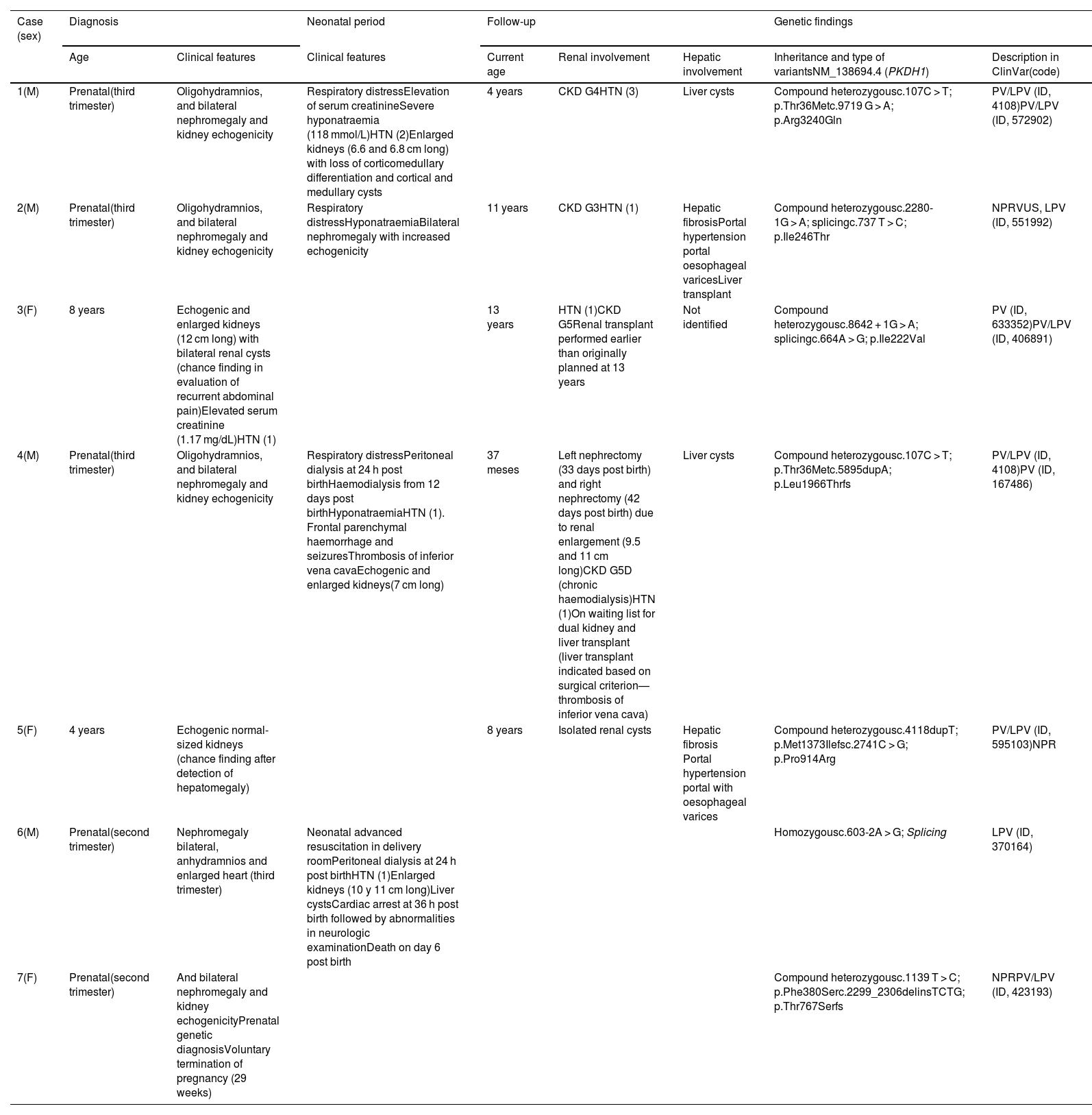
Table 1 presents the clinical characteristics at baseline and during the follow-up and presents the pathogenic variants of the PKHD1 gene detected in the 7 patients. As would be expected in a sample including recently diagnosed cases, most patients (5/7) received the diagnosis antenatally, although only 2 cases were diagnosed as early as the second trimester of gestation, the period when diagnosis is most common at the time of this writing. The clinical manifestations in patients in our case series reflected the substantial phenotypic heterogeneity of ARPKD, including late termination of pregnancy, severe or even fatal respiratory and renal complications that required complex treatment approaches in the early postnatal period, mild and slowly progressing impairment of renal function and even chance diagnosis during childhood.
Clinical features and pathogenic variants of the PKHD1 gene in the cases of autosomal recessive polycystic kidney disease included in the study. In cases with hypertension (HTN), the number of drugs used to control it is indicated in parentheses.
| Case (sex) | Diagnosis | Neonatal period | Follow-up | Genetic findings | ||||
|---|---|---|---|---|---|---|---|---|
| Age | Clinical features | Clinical features | Current age | Renal involvement | Hepatic involvement | Inheritance and type of variantsNM_138694.4 (PKDH1) | Description in ClinVar(code) | |
| 1(M) | Prenatal(third trimester) | Oligohydramnios, and bilateral nephromegaly and kidney echogenicity | Respiratory distressElevation of serum creatinineSevere hyponatraemia (118 mmol/L)HTN (2)Enlarged kidneys (6.6 and 6.8 cm long) with loss of corticomedullary differentiation and cortical and medullary cysts | 4 years | CKD G4HTN (3) | Liver cysts | Compound heterozygousc.107C > T; p.Thr36Metc.9719 G > A; p.Arg3240Gln | PV/LPV (ID, 4108)PV/LPV (ID, 572902) |
| 2(M) | Prenatal(third trimester) | Oligohydramnios, and bilateral nephromegaly and kidney echogenicity | Respiratory distressHyponatraemiaBilateral nephromegaly with increased echogenicity | 11 years | CKD G3HTN (1) | Hepatic fibrosisPortal hypertension portal oesophageal varicesLiver transplant | Compound heterozygousc.2280-1G > A; splicingc.737 T > C; p.Ile246Thr | NPRVUS, LPV (ID, 551992) |
| 3(F) | 8 years | Echogenic and enlarged kidneys (12 cm long) with bilateral renal cysts (chance finding in evaluation of recurrent abdominal pain)Elevated serum creatinine (1.17 mg/dL)HTN (1) | 13 years | HTN (1)CKD G5Renal transplant performed earlier than originally planned at 13 years | Not identified | Compound heterozygousc.8642 + 1G > A; splicingc.664A > G; p.Ile222Val | PV (ID, 633352)PV/LPV (ID, 406891) | |
| 4(M) | Prenatal(third trimester) | Oligohydramnios, and bilateral nephromegaly and kidney echogenicity | Respiratory distressPeritoneal dialysis at 24 h post birthHaemodialysis from 12 days post birthHyponatraemiaHTN (1). Frontal parenchymal haemorrhage and seizuresThrombosis of inferior vena cavaEchogenic and enlarged kidneys(7 cm long) | 37 meses | Left nephrectomy (33 days post birth) and right nephrectomy (42 days post birth) due to renal enlargement (9.5 and 11 cm long)CKD G5D (chronic haemodialysis)HTN (1)On waiting list for dual kidney and liver transplant (liver transplant indicated based on surgical criterion— thrombosis of inferior vena cava) | Liver cysts | Compound heterozygousc.107C > T; p.Thr36Metc.5895dupA; p.Leu1966Thrfs | PV/LPV (ID, 4108)PV (ID, 167486) |
| 5(F) | 4 years | Echogenic normal-sized kidneys (chance finding after detection of hepatomegaly) | 8 years | Isolated renal cysts | Hepatic fibrosis Portal hypertension portal with oesophageal varices | Compound heterozygousc.4118dupT; p.Met1373Ilefsc.2741C > G; p.Pro914Arg | PV/LPV (ID, 595103)NPR | |
| 6(M) | Prenatal(second trimester) | Nephromegaly bilateral, anhydramnios and enlarged heart (third trimester) | Neonatal advanced resuscitation in delivery roomPeritoneal dialysis at 24 h post birthHTN (1)Enlarged kidneys (10 y 11 cm long)Liver cystsCardiac arrest at 36 h post birth followed by abnormalities in neurologic examinationDeath on day 6 post birth | Homozygousc.603-2A > G; Splicing | LPV (ID, 370164) | |||
| 7(F) | Prenatal(second trimester) | And bilateral nephromegaly and kidney echogenicityPrenatal genetic diagnosisVoluntary termination of pregnancy (29 weeks) | Compound heterozygousc.1139 T > C; p.Phe380Serc.2299_2306delinsTCTG; p.Thr767Serfs | NPRPV/LPV (ID, 423193) | ||||
Additional pathogenic or likely pathogenic variants were not detected in any other of the analysed genes.
ClinVar: database of the National Library of Medicine documenting the association between human genes and phenotypes (ncbi.nlm.nih.gov/clinvar). CKD, chronic kidney disease (G, CKD stage); F, female; HTN, hypertension; LPV, likely pathogenic variant; M, male; NPR, not previously reported; PV, pathogenic variant; VUS, variant of uncertain significance.
When it came to diagnosis, in agreement with the previous literature, the presence of bilateral enlarged echogenic kidneys and oligohydramnios was the presentation most frequently leading to prenatal detection of ARPKD.2 However, we ought to highlight two clinically relevant aspects deduced from the features observed in our patients, on one hand, while kidney enlargement is found in nearly every case, the presence of normally sized kidneys does not rule out the disease (patient 5 had kidneys of normal size at age 8 years); on the other, the presence of renal cysts detectable by ultrasound is not a common feature in cases diagnosed in the perinatal period, which may give rise to diagnostic uncertainty if the clinician and/or radiologists are not acquainted with the disease.
Our findings also corroborated the strong association between prenatal nephromegaly and oligohydramnios and greater postnatal disease severity. Along with other renal and nutritional problems, this association also seems to be closely related to respiratory complications,2 secondary to pulmonary hypoplasia, pneumothorax or abdominal distention itself, which may be fatal in up to 30% of cases,3 as occurred in patient 6 in our series.
Among the less frequently described clinical aspects of ARPKD, although they are not rare, given their presence in this case series, we ought to highlight the low frequency of surgical management with nephrectomy, an approach that is usually indicated due to the negative impact of nephromegaly on nutrition and respiratory function4 and accelerated renal growth in the early postnatal period, both of which were present in patient 4 (kidneys measuring 9.5 and 11 cm at 1 month post birth). While this uncontrolled rapid growth may occur simultaneously and proportionally in both kidneys, it is even more frequent following unilateral nephrectomy, which can then hasten the need for a second nephrectomy, something that also was the case in the same patient, a decision that was made after thorough consideration, given the poor long-term neurologic outcomes that have been reported in the previous literature.5
Another two postnatal manifestations that have been thoroughly documented in the literature and corroborated in our case series are hyponatraemia and hypertension, of which the underlying mechanisms have yet to be clearly established.6 Hyponatraemia is usually asymptomatic and tends to resolve within a few months, although it can also be severe, as was the case of patient 1 (trough sodium level of 118 mmol/L). Hypertension, however, may be associated with severe cardiovascular events (brain haemorrhage in patient 4), frequently requires the use of multiple drugs and may persist for a long time, features that were also observed in other patients.
The published evidence on the genotype-phenotype correlation in patients with ARPKD is not consistent. In our series, patient 6, who carried a homozygous splicing variant, had the worst outcome, dying 6 days after birth. The combination of 2 pathogenic splicing variants is associated with more severe renal impairment and an increased probability of developing portal hypertension. However, the severity of disease cannot be attributed exclusively to the genetic background, as up to 30% of affected infants die in the neonatal period or within 1 year post birth, but chiefly on account of associated respiratory disorders. The (NM_138694.4),c.107C>T; p.Thr36Met missense variant of the PKHD1 gene, which is the one most frequently reported in patients with ARPKD, was found in compound heterozygosis with two different types of variants in 2 cases in our series (patients 1 and 4). Although we cannot attribute greater severity to its presence, we ought to highlight that both of these patients had an early prenatal onset of disease.
In brief, while we present a small number of cases, the clinical manifestations observed in our patients allowed confirmation of the current predominance of prenatal diagnosis and the broad phenotypic spectrum of ARPKD, within which respiratory complications pose a substantial therapeutic challenge in the neonatal period. The potential association with a normal kidney size at onset or accelerated postnatal renal growth, which may warrant consideration of nephrectomy, are aspects that have not been reported as frequently in the past. Advances in the knowledge of ARPKD require the performance of collaborative studies of larger scope to be able to establish with greater accuracy the association between clinical manifestations and the described pathogenic variants of the PKHD1 gene and other genes associated with this disease.
FundingThis research did not receive any external funding.
