


Neonatal cystic fibrosis (CF) screening has enabled the disease to be diagnosed early, and is a determining factor in the increase in survival of these patients. Its main disadvantage is its low specificity and elevated number of false positives. The aim of this study is to analyse the differences in immunoreactive trypsin (IRT) between the different groups of newborns (NB) with a positive neonatal screen depending on whether they were healthy, healthy carriers, affected by CF, or CFSPID (Cystic Fibrosis Screen Positive, Inconclusive Diagnosis).
MaterialRetrospective analytical study of the concentrations of IRT in NB with a positive neonatal screen for CF born in a tertiary hospital during an 8-year period.
ResultsA total of 790 NB with a positive neonatal screen for CF were analysed. Of these 86.3% were term, 53% girls, and 11.8% were admitted. The mean IRT value was 79.16 ng/mL (range 60–367). Significantly higher concentrations of IRT were found in those affected by CF compared to the other groups (P < .001). There were higher levels in large prematures (P = .007) and admitted patients (P = .002). There were no differences as regards gender or season. There was a direct correlation of 64% (P = .001) between IRT and sweat test in those affected by CF and CFSPID. A cut-off value of IRT for the diagnosis of CF was calculated from the ROC curve (76.2 ng/mL (S = 95.7%, Sp = 64.5%).
ConclusionsNB with CF have significantly higher levels of IRT than healthy ones, or carriers and CFSPID. Prematurity and hospital admission may also have an influence. A higher IRT value is associated with a higher level in the sweat test.
El cribado neonatal de fibrosis quística (FQ) ha permitido el diagnóstico precoz de la enfermedad, siendo determinante en el aumento de supervivencia de estos pacientes. Su principal inconveniente es su baja especificidad y elevado número de falsos positivos. El objetivo fue analizar las diferencias de tripsina inmunorreactiva (TIR) entre los diferentes grupos de recién nacidos (RN) con cribado neonatal positivo según fueran sanos, portadores sanos, afectos de FQ o Cystic Fibrosis Screen Positive, Inconclusive Diagnosis (CFSPID).
MaterialEstudio retrospectivo analítico de las concentraciones de TIR en RN con cribado neonatal positivo para FQ nacidos en un hospital de tercer nivel durante 8 años.
ResultadosSe analizaron 790 RN con cribado neonatal positivo para FQ, 86,3% a término, 53% niñas y 11,8% ingresados. El valor medio de TIR fue 79,16 ng/mL (rango 60–367). Se encontraron concentraciones significativamente más elevadas de TIR en afectos de FQ con respecto a los otros grupos (p < 0,001). Existen niveles superiores en grandes prematuros (p = 0,007) e ingresados (p = 0,002). No difieren en cuanto a sexo o estacionalidad. Existe una correlación directa del 64% (p = 0,001) entre TIR y test del sudor en afectos de FQ y CFSPID. Mediante curva ROC se calculó el valor de corte de TIR para el diagnóstico de FQ, que fue 76,2 ng/mL (S = 95,7%, E = 64,5%).
ConclusionesLos RN con FQ presentan cifras significativamente más elevadas de TIR que sanos, portadores o CFSPID. La prematuridad y hospitalización también pueden influir. Un mayor valor de TIR se relaciona con una mayor cifra en el test del sudor.
Cystic fibrosis (CF) is the most prevalent severe autosomal recessive genetic disorder in Caucasian populations. Its incidence in Spain is estimated at 1 case per 5000 live births, while 1 in 35 inhabitants are healthy carriers of the disease.
The incidence of this disease and the availability of a relatively simple, feasible and highly sensitive screening method largely justifies the institution of a newborn screening programme.
One of the advantages of screening is early diagnosis, which allows early treatment of the disease. As a result, the survival and life expectancy of patients with CF have increased spectacularly in recent years. Based on the most recent data of the Cystic Fibrosis Foundation Patient Registry, from 2018, the median life expectancy at present is 47.4 years.1 This is partly due to the widespread domestic and international implementation of newborn CF screening programmes in recent years2 and to improvements in treatment and care in the past few decades.3–5
In 1979 Crossley et al. found that immunoreactive trypsinogen (IRT) levels were elevated in blood samples obtained from children with CF. The elevation of IRT in blood results from the partial or total obstruction of the exocrine ducts of the pancreas leading to “back-leakage” of the enzyme into the plasma.6 After publication of this article, measurement of IRT in a dry blood spot became the gold standard for screening and is currently included in most current protocols to be performed between days 3 and 5 post birth.
The cut-off value is a crucial aspect of screening and varies between programmes from the 95th to the 99th percentile, with lower thresholds offering a higher sensitivity but less specificity.7 It is important to take into consideration that the concentration of IRT varies depending on the age of the neonate and decreases substantially from week 3 post birth.
Different strategies and protocols have been established for newborn CF screening, including the following:
- •
IRT/IRT protocol: includes a second measurement of IRT between 25 and 40 days post birth, and if this second test is positive, genetic testing is performed.
- •
IRT/DNA protocol: after detection of elevated IRT levels, genetic testing is performed in the same heel prick capillary blood sample, analysing the variants that are most prevalent locally. This IRT/DNA strategy, which is followed by performance of the sweat chloride test, is the one used most frequently in the Autonomous Community of Aragon and the strategy evaluated in our study. Since 2011, the Elucigene® assay (Elucigene Diagnostics, Manchester, United Kingdom) is the test used to screen for 50 variants in the CFTR gene. In case the initial genetic screening detects changes in any of the alleles or the results of the sweat chloride test are inconclusive or abnormal, genetic testing is expanded with collection of a peripheral blood sample from the infant and the parents.8
However, the diagnosis of CF becomes complex as the number of described CFTR variants increases and the limits that define patient subgroups blur. The detection of variants of uncertain significance poses a challenge to diagnosis and to the interpretation of inconclusive newborn screening results. To reflect this, the European Cystic Fibrosis Society Neonatal Screening Working Group recently proposed the term “cystic fibrosis screen positive, inconclusive diagnosis” (CFSPID), which includes: neonates with 2 variants in the CFTR gene (with only one of them known to cause CF) and a normal sweat Cl concentration, and neonates with one or no variant in the CFTR gene and an intermediate sweat Cl concentration.8–10 In addition, this screening approach generates a substantial number of false positives.11,12 Several studies have suggested that elevated levels of IRT are associated with the Cl concentration found in the sweat chloride test and are equally affected by the genotype.13
Considering the above, the aim of our study was to address the need to identify factors influencing the level of IRT and that could be associated with false positive results. We also sought to establish the optimal cut-off point to detect patients with CF allowing earlier intervention and management.
The primary objective of the study was to analyse the differences in IRT levels between different groups of neonates with a positive newborn screening result for CF based on whether they were healthy, healthy carriers, CFSPID or had CF. The secondary objectives were to analyse the potential correlation between IRT values with the variables under study (sex, gestational age, sweat chloride test, neonatal admission and genotype), assess the predictive value of IRT and establish a cut-off value for IRT with a greater positive predictive value for detection of classic CF.
Material and methodsWe performed a retrospective inferential study in neonates with a positive newborn screening result for CF, defined as an IRT level of 60 ng/mL or greater.
For the 2011–2019 period, we included infants with abnormal newborn screening results born in a tertiary care hospital and the patients with CF or CFSPID managed in the same hospital, independently of the year and place of birth.
We excluded infants born at term or preterm that underwent newborn screening after week 3 post birth and infants with a positive screening result in whom the sweat chloride test was not performed or the result of this test was not documented in the health record
We collected data on the following clinical variables: decimal age, sex, birth weight (g), weeks of gestation, hospital admission in the neonatal, IRT levels (ng/mL), NaCl conductivity in the sweat test (mEq/L) and genotype.
The variants included in the analysis were: R347H, R347P, 2789+5G>A, 3120+1G>A, 711+1G>T, R334W, I507del, F508del, 3849+10KbC>T, 1677delTA, 1078delT, V520F, L206W, W1282X, R560T, 2347delG, Q890X, R553X, G551D, S549R(T>G), S549N, M1101K, G542X, 3905insT, Y1092X(C>A), S1251N, 444delA, 1811+1.6kbA>G, 1717-1G>A, R117H, R117C, N1303K, Y122X, 394delTT, G85E, R1066C, 1898+1G>A, W846X, 2184delA, D1152H, CFTRdele2.3, P67L, 2143delIT, E60X, 3659delC, 3272-26A>G, 621+1G>T, A455E, R1162X, R1158×. We also analysed the poly(T) tail of intron 8 (5T, 7T, 9T) and the adjacent (TG)n motif.
Participants were classified into subgroups based on gestational age, defining very preterm (VPT) as birth before 32 weeks’ gestation, late preterm (LPT) as birth between 32 and 37 weeks’ gestation and full term (FT) as birth from 37 weeks’ gestation, and also based on the results of newborn screening for CF as healthy, healthy carrier, CFSPID or CF (Table 1).
Classification of neonates by subgroups.
| Neonate subgroups | |
|---|---|
| 1. Healthy | No variants and normal sweat chloride value |
| 2. Healthy carrier | One variant and normal sweat chloride value |
| 3. CFSPID | Two variants, only one classified as CF-causinga, with normal sweat chloride value |
| One or no variant with inconclusive sweat chloride test | |
| 4. CF | Two CF-causing variants and/or abnormal sweat chloride value |
CF, cystic fibrosis; CFSPID, cystic fibrosis screen positive, inconclusive diagnosis.
We performed the statistical analysis with the software IBM SPSS Statistics Version 15.0, choosing an alpha level of 5%.
ResultsBetween January 2011 and September 2019, there were a total of 790 neonates with a positive screening result for CF.
The distribution by gestational age was 682 (86.3%) FT, 43 (5.4%) LPT and 25 (3.2%) VPT born before 32 weeks’ gestation, and the range of gestational age at birth was 24–42 weeks. Fifty-three percent were female and the mean birth weight was 3083 g with a range of 400–4840 g. The proportion admitted to hospital during the neonatal period was 11.8%.
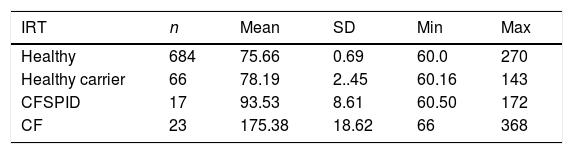
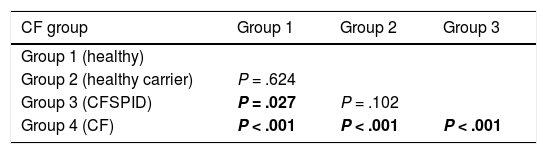
The mean IRT level was 79.16 ng/mL (range, 60–367). Table 2 presents the mean levels of IRT in each subgroup of patients, with a mean of 75.66 ng/mL in healthy infants, 78.19 ng/mL in healthy carriers, 93.53 ng/mL in CFSPID cases and 175.82 ng/mL in CF cases. We found significantly higher levels of IRT in cases of CF (P < .001) compared to cases of CFSPID, healthy carriers and healthy infants (Table 3, Fig. 1).
Mean, standard deviation and range of IRT values (ng/mL) by patient subgroup.
| IRT | n | Mean | SD | Min | Max |
|---|---|---|---|---|---|
| Healthy | 684 | 75.66 | 0.69 | 60.0 | 270 |
| Healthy carrier | 66 | 78.19 | 2..45 | 60.16 | 143 |
| CFSPID | 17 | 93.53 | 8.61 | 60.50 | 172 |
| CF | 23 | 175.38 | 18.62 | 66 | 368 |
CF, cystic fibrosis; CFSPID, cystic fibrosis screen positive, inconclusive diagnosis; IRT, immunoreactive trypsinogen; SD, standard deviation.
Comparison of IRT levels in different groups of patients.
| CF group | Group 1 | Group 2 | Group 3 |
|---|---|---|---|
| Group 1 (healthy) | |||
| Group 2 (healthy carrier) | P = .624 | ||
| Group 3 (CFSPID) | P = .027 | P = .102 | |
| Group 4 (CF) | P < .001 | P < .001 | P < .001 |
Statistically significant differences between groups shown in boldface.
CF, cystic fibrosis; CFSPID, cystic fibrosis screen positive, inconclusive diagnosis; IRT, immunoreactive trypsinogen.
 by patient subgroup. CF, cystic fibrosis; CFSPID, cystic fibrosis screen positive, inconclusive diagnosis; IRT, immunoreactive trypsinogen.")
We also found higher levels of IRT in infants born preterm (P = .007). We observed higher levels of IRT in the VPT group (median, 78.35 ng/mL) compared to the LPT group (median, 73 ng/mL) and the FT group (70.12 ng/mL), differences that were statistically significant.
We found significant differences in IRT values in infants admitted to hospital in the neonatal period compared to infants that were not admitted (n = 93 vs n = 586; P = .002). The mean IRT value in the admitted group was 84.50 ng/mL (95% CI, 78.23–90.77) compared to 78.15 ng/mL in the not-admitted group (95% CI, 75.69–80.62). The difference was greater in infants admitted to the intensive care unit.
We did not find differences in IRT levels based on sex or season.
The mean conductivity of NaCl in sweat was 30.56 mmol/L (range, 13−270 mmol/L). We found a direct correlation of 64% (P = .001) between the IRT levels and the sweat test results in patients with CF and CFSPID.
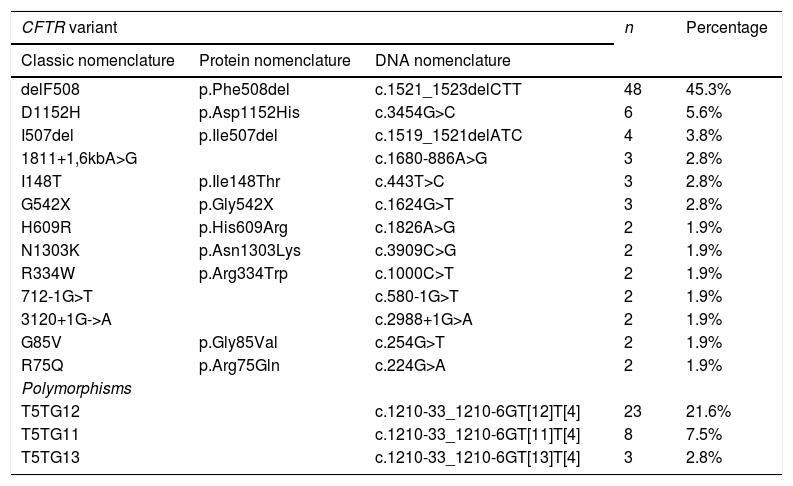
When it came to DNA testing, 684 infants (86.6%) had no changes in the CFTR gene. The most frequent variant was the one classically known as delF508, found in heterozygosis in 40 patients (5.06%) and in homozygosis in 8 (0.9%). Table 4 presents the most frequent variants and polymorphisms in the total of patients with abnormal genetic tests results (including healthy carriers and patients with CFSPID and CF).
Prevalence of the most frequent CFTR gene variants in 1 or 2 alleles (n = 160).
| CFTR variant | n | Percentage | ||
|---|---|---|---|---|
| Classic nomenclature | Protein nomenclature | DNA nomenclature | ||
| delF508 | p.Phe508del | c.1521_1523delCTT | 48 | 45.3% |
| D1152H | p.Asp1152His | c.3454G>C | 6 | 5.6% |
| I507del | p.Ile507del | c.1519_1521delATC | 4 | 3.8% |
| 1811+1,6kbA>G | c.1680-886A>G | 3 | 2.8% | |
| I148T | p.Ile148Thr | c.443T>C | 3 | 2.8% |
| G542X | p.Gly542X | c.1624G>T | 3 | 2.8% |
| H609R | p.His609Arg | c.1826A>G | 2 | 1.9% |
| N1303K | p.Asn1303Lys | c.3909C>G | 2 | 1.9% |
| R334W | p.Arg334Trp | c.1000C>T | 2 | 1.9% |
| 712-1G>T | c.580-1G>T | 2 | 1.9% | |
| 3120+1G->A | c.2988+1G>A | 2 | 1.9% | |
| G85V | p.Gly85Val | c.254G>T | 2 | 1.9% |
| R75Q | p.Arg75Gln | c.224G>A | 2 | 1.9% |
| Polymorphisms | ||||
| T5TG12 | c.1210-33_1210-6GT[12]T[4] | 23 | 21.6% | |
| T5TG11 | c.1210-33_1210-6GT[11]T[4] | 8 | 7.5% | |
| T5TG13 | c.1210-33_1210-6GT[13]T[4] | 3 | 2.8% | |
The absolute frequency and percentage values correspond to the number of patients carrying the variant in one or both alleles. The table does not include variants with a percentage of less than 1%.
CFTR: cystic fibrosis transmembrane conductance regulator protein.
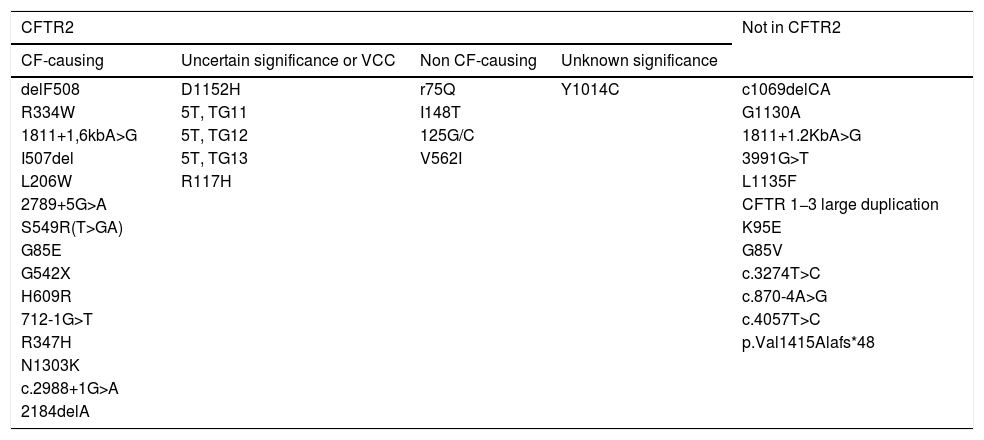
We classified the detected variants according to the definitions established in the Clinical and Functional Translation of CFTR database (CFTR2) (Table 5).14 In the subgroup of patients with CF and CFSPID, we found significantly higher levels of IRT in patients with 2 CF-causing variants CF (n = 16), with a mean IRT level of 190.86 ng/mL, compared to patients with a single CF-causing variant and one non CF-causing variant (n = 4), with a mean level of 70.22 ng/mL (P = .003) or patients with one CF-causing variant and one variant of unknown significance (n = 7), with a mean level of 108.24 ng/mL (P = .027).
Classification of the identified mutations based on the variants included in the CFTR2 database.
| CFTR2 | Not in CFTR2 | |||
|---|---|---|---|---|
| CF-causing | Uncertain significance or VCC | Non CF-causing | Unknown significance | |
| delF508 | D1152H | r75Q | Y1014C | c1069delCA |
| R334W | 5T, TG11 | I148T | G1130A | |
| 1811+1,6kbA>G | 5T, TG12 | 125G/C | 1811+1.2KbA>G | |
| I507del | 5T, TG13 | V562I | 3991G>T | |
| L206W | R117H | L1135F | ||
| 2789+5G>A | CFTR 1−3 large duplication | |||
| S549R(T>GA) | K95E | |||
| G85E | G85V | |||
| G542X | c.3274T>C | |||
| H609R | c.870-4A>G | |||
| 712-1G>T | c.4057T>C | |||
| R347H | p.Val1415Alafs*48 | |||
| N1303K | ||||
| c.2988+1G>A | ||||
| 2184delA | ||||
CFTR2, The Clinical and Functional Translation of CFTR, available at http://cftr2.org; VCC, varying clinical consequence.
In order to establish the optimal cut-off point, we generated a receiver operating characteristic (ROC) curve with an area under the curve of 0.93 (P < .001). Using the ROC curve, we obtained a cut-off value of IRT for prediction of CF of 76.2 ng/mL, with a sensitivity of 95.7% and a specificity of 64.5%.
DiscussionThe recent introduction of newborn CF screening through the measurement of IRT levels has resulted in significant improvements in the survival and quality of life of affected patients by allowing early diagnosis of the disease.15 Still, this is a screening test and as such has a low specificity, which translates to a high number of false positive results.
The main disadvantage that results from its low specificity is the anxiety it produces in families and the high cost of performing DNA testing and sweat chloride tests in every infant with a positive result. On the other hand, it allows diagnosis in healthy carriers, individuals with atypical forms of CF and even individuals with variants of uncertain significance,16,17 whose inclusion in the indication for diagnostic testing is currently under debate due to the lesser benefits derived from diagnosis and the greater anxiety generated in families.
Given the above, the importance of the selection of the cut-off point for IRT levels in each hospital becomes evident.
We found a mean IRT level of 79.16 ng/mL and a maximum of 367 ng/mL in one patient with CF that carried two severe variants.
When it came to the sweat chloride test, most measurements (96.1%) were normal, that is, below the established threshold of 50 mmol/L, with a mean value of 30.56 mmol/L.
In the analysis by subset, we found significantly greater levels of IRT in the CF group (P < .001) compared to the healthy infant and healthy carrier groups, as was the case with the CFSPID group. We found differences between the CFSPID and healthy infant group (P = .027). These results were similar to and consistent with those of a study conducted Paracchini et al., who concluded that higher levels of IRT in a heel prick blood sample are associated with an increased probability of CF.18 As occurred in our study, these authors did not find differences between healthy infants and healthy carriers.
However, some studies by Castellani et al. found significant differences between healthy children and healthy carriers.19 While we did not observe this in the sample under study, we did find a tendency toward higher IRT levels in the carrier group compared to the healthy group. However, the distribution of IRT levels overlapped substantially between the 4 groups and especially between the first 3 (healthy, healthy carriers and CFSPID). Therefore, the predictive power of IRT levels for categorization of these patients is low. On the other hand, the absence of a significant association in our study may be due to the lower sample size compared to those of the aforementioned studies.18,19
Preterm birth may be independently associated with IRT elevation, as proposed by Korzeniewsli et al., who found significantly higher levels of IRT in neonates with very low birth weight (<1500 g) and born extremely preterm (gestational age <28 weeks).20 This is consistent with our findings, as IRT levels were higher in the VPT group compared to the LPT group (P = .008) and the FT group (P = .002).
Stressful birth and birth asphyxia have been described as causes of IRT elevation,21 which could be related to the differences we observed based on the history of neonatal admission. A study by Rock et al. found significantly lower Apgar scores at 1 and 5 min in patients with elevated IRT compared to the general population.22 In their analysis, the mean IRT level in hospitalised neonates was greater compared to neonates that did not require admission, and higher in infants admitted to the neonatal intensive care unit (NICU), although the latter may have to do with most infants admitted to the NICU having been born LPT.
In agreement with the reports of other authors,9 there was a direct correlation between IRT levels and sweat test values, with a stronger correlation in patients with CF and CFSPID (r = 0.64; P = .001), corresponding to a linear correlation of 64% between these two variables.
In patients with abnormal results in the DNA test, the most frequently detected variant was delF508 (45%). We ought to highlight the substantial molecular heterogeneity of the sample under study, as no other variant had a frequency greater than 5% (Table 4).
Patients with 2 CF-causing variants had significantly higher levels of IRT. These results were similar to those described in other case series,23,24 with reported lower levels of IRT in blood and NaCl in sweat in patients with a non CF-causing variant or a rare variant compared to patients with 2 CF-causing variants.
In our local population, the optimal cut-off value for diagnosis of CF would be 76.2 ng/mL, with a sensitivity of 95.7% and a specificity of 64.5%, with serum IRT levels above this value corresponding to a greater probability of CF.
In any case, we think it prudent not to raise IRT cut-off value used for detection of CF in the screening, as carriers and patients with atypical forms would not be identified, while using this IRT level only as a marker to facilitate early diagnosis. This would be particularly useful in patients with a single variant detected in the initial genetic test that has yet to undergo comprehensive genetic testing, with the probability of additional variants being greater in patients with very elevated levels of IRT.
One of the limitations of the study is its observational design with retrospective data collection. On the other hand, its main strength is the large sample size for a study conducted in a single hospital. Additional multicentre studies with prospective data collection are required to corroborate these findings.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Arrudi-Moreno M, García-Romero R, Samper-Villagrasa P, Sánchez-Malo MJ, Martin-de-Vicente C. Cribado neonatal de fibrosis quística: análisis y diferencias de los niveles de tripsina inmunorreactiva en recién nacidos con cribado positivo. An Pediatr (Barc). 2021;95:11–17.
