



El cribado neonatal de fibrosis quística (FQ) ha permitido el diagnóstico precoz de la enfermedad, siendo determinante en el aumento de supervivencia de estos pacientes. Su principal inconveniente es su baja especificidad y elevado número de falsos positivos. El objetivo fue analizar las diferencias de tripsina inmunorreactiva (TIR) entre los diferentes grupos de recién nacidos (RN) con cribado neonatal positivo según fueran sanos, portadores sanos, afectos de FQ o Cystic Fibrosis Screen Positive, Inconclusive Diagnosis (CFSPID).
MaterialEstudio retrospectivo analítico de las concentraciones de TIR en RN con cribado neonatal positivo para FQ nacidos en un hospital de tercer nivel durante 8 años.
ResultadosSe analizaron 790RN con cribado neonatal positivo para FQ, 86,3% a término, 53% niñas y 11,8% ingresados. El valor medio de TIR fue 79,16ng/ml (rango 60-367). Se encontraron concentraciones significativamente más elevadas de TIR en afectos de FQ con respecto a los otros grupos (p<0,001). Existen niveles superiores en grandes prematuros (p=0,007) e ingresados (p=0,002). No difieren en cuanto a sexo o estacionalidad. Existe una correlación directa del 64% (p=0,001) entre TIR y test del sudor en afectos de FQ y CFSPID. Mediante curva ROC se calculó el valor de corte de TIR para el diagnóstico de FQ, que fue 76,2ng/ml (S=95,7%, E=64,5%).
ConclusionesLos RN con FQ presentan cifras significativamente más elevadas de TIR que sanos, portadores o CFSPID. La prematuridad y hospitalización también pueden influir. Un mayor valor de TIR se relaciona con una mayor cifra en el test del sudor.
Neonatal cystic fibrosis (CF) screening has enabled the disease to be diagnosed early, and is a determining factor in the increase in survival of these patients. Its main disadvantage is its low specificity and elevated number of false positives. The aim of this study is to analyse the differences in immunoreactive trypsin (IRT) between the different groups of newborns (NB) with a positive neonatal screen depending on whether they were healthy, healthy carriers, affected by CF, or CFSPID (Cystic Fibrosis Screen Positive, Inconclusive Diagnosis).
MaterialRetrospective analytical study of the concentrations of IRT in NB with a positive neonatal screen for CF born in a tertiary hospital during an 8-year period
ResultsA total of 790 NB with a positive neonatal screen for CF were analysed. Of these 86.3% were term, 53% girls, and 11.8% were admitted. The mean IRT value was 79.16 ng/mL (range 60 – 367). Significantly higher concentrations of IRT were found in those affected by CF compared to the other groups (P<.001). There were higher levels in large prematures (P=.007) and admitted patients (P=.002). There were no differences as regards gender or season. There was a direct correlation of 64% (P=.001) between IRT and sweat test in those affected by CF and CFSPID. A cut-off value of IRT for the diagnosis of CF was calculated from the ROC curve (76.2 ng/mL (S=95.7%, Sp=64.5%).
ConclusionsNB with CF have significantly higher levels of IRT than healthy ones, or carriers and CFSPID. Prematurity and hospital admission may also have an influence. A higher IRT value is associated with a higher level in the sweat test.
La fibrosis quística (FQ) es la enfermedad genética grave, de herencia autosómica recesiva, más frecuente en las poblaciones de origen caucásico. Su incidencia en nuestro entorno se estima en torno a un caso de cada 5.000 nacidos vivos, mientras que uno de cada 35 habitantes son portadores sanos de la enfermedad.
La incidencia de esta enfermedad junto con un método de cribado relativamente simple y fácil de realizar y con una alta sensibilidad justifica, en gran medida, la implantación de un programa de cribado neonatal.
Una de las ventajas del cribado ha sido permitir un diagnóstico temprano que condicione un tratamiento precoz de la enfermedad. De esta manera, el pronóstico de supervivencia en pacientes afectos de FQ ha aumentado espectacularmente en los últimos años. De acuerdo con los últimos datos del registro de Cystic Fibrosis Foundation de 2018, la mediana de supervivencia actual es de 47,4años1. Esto es, en parte, debido a la implantación de los programas de cribado neonatal de FQ que se han generalizado en el territorio nacional e internacional en los últimos años2, así como a las mejoras en los tratamientos y la atención en las últimas décadas3–5.
En 1979 Crossley et al. demostraron que los valores de tripsina inmunorreactiva (TIR) estaban elevados en las muestras de sangre obtenidas en los niños afectos de FQ. La concentración elevada de TIR en sangre es consecuencia de la obstrucción parcial o completa de los canalículos pancreáticos que produce que refluya dicha enzima hacia la circulación sanguínea6. Tras esta publicación, la determinación de la TIR en una gota de sangre seca se impuso como método definitivo y está contemplada su realización, en la mayoría de los protocolos actuales, entre el tercer y quinto día de vida.
El establecimiento del punto de corte es un punto crítico y varía según los programas entre el percentil 95 al 99, considerando los valores bajos más sensibles pero menos específicos7. Hay que tener en cuenta que la concentración de TIR es dependiente de la edad del recién nacido (RN), disminuyendo notablemente a partir de la tercera semana de vida.
Se han establecido diferentes estrategias y protocolos de cribado neonatal de FQ, entre los cuales están:
- •
Protocolo TIR/TIR: incluyen una segunda determinación de TIR entre los 25-40 días de vida, y si esta segunda muestra continúa elevada se realiza el estudio genético.
- •
Protocolo TIR/ADN: tras una determinación de TIR elevada se realiza un estudio genético de la misma sangre de talón con la determinación de las mutaciones con mayor prevalencia en nuestro medio. Esta estrategia TIR/ADN, con posterior realización del test del sudor, es la empleada en la Comunidad Autónoma de Aragón y la analizada en este estudio. Desde 2011 se utiliza el sistema de análisis Elucigene® (Elucigene Diagnostics, Manchester, Reino Unido) para el rastreo de 50 mutaciones en el gen CFTR. En el caso que el estudio genético inicial presente una mutación en alguno de los alelos y/o presente un test del sudor dudoso o alterado, se amplía el estudio genético mediante la extracción de sangre periférica en el niño y sus progenitores8.
Sin embargo, el diagnóstico de la enfermedad a medida que aumenta el número de mutaciones en el gen CFTR descritas es complejo, y los límites que definen los subgrupos afectos se difuminan. La aparición de mutaciones de significado incierto supone un reto en el diagnóstico y en la interpretación de resultados no concluyentes en el cribado neonatal. En este sentido, el grupo de trabajo europeo de cribado neonatal de FQ ha propuesto recientemente el término Cystic Fibrosis Screen Positive, Inconclusive Diagnosis (CFSPID) que incluye: RN con 2 mutaciones en CFTR (solo una de ellas causante de FQ) y valores de Cl en sudor normales y RN con una o ninguna mutación en CFTR y valores intermedios de Cl en el sudor8–10. Además, este sistema de cribado conduce a un número apreciable de falsos positivos11,12. Diversos estudios han sugerido que concentraciones elevadas de TIR se relacionan con los niveles obtenidos en el test del sudor y están igualmente influenciados por el genotipo13.
Por todo ello se plantea en el presente estudio la necesidad de establecer factores que influyan en la concentración de TIR y que puedan condicionar «falsos positivos». Del mismo modo, determinar un punto de corte que permita detectar con mayor probabilidad pacientes afectos de FQ y una actuación y seguimiento más precoces.
El objetivo principal de este trabajo fue analizar las diferencias en los niveles de TIR entre los diferentes grupos de RN con cribado positivo para FQ según fueran sanos, portadores sanos, CFSPID o afectos de FQ. Como objetivos secundarios se analizó la posible correlación entre los valores de TIR en función de las variables analizadas (sexo, edad gestacional, test del sudor, hospitalización en periodo neonatal y genotipo), evaluar el valor predictivo de TIR y determinar un punto de corte de TIR con mayor valor predictivo positivo para FQ clásica.
Material y métodosSe realizó un estudio analítico retrospectivo en RN con cribado neonatal positivo para FQ, definido como cifras de TIR igual o por encima de 60ng/ml.
Se incluyeron durante el periodo de tiempo comprendido entre 2011-2019: RN con cribado neonatal alterado nacidos en un hospital de tercer nivel y pacientes afectos de FQ y CFSPID controlados en dicho hospital durante el periodo del estudio, independientemente del año y de su lugar de nacimiento.
Como criterios de exclusión: RN a término o prematuros con cribado realizado después de la tercera semana de vida y RN con cribado positivo a los que no se haya realizado test del sudor o no consten datos.
Se recogieron las siguientes variables clínicas: edad decimal, sexo, peso al nacimiento (g), semanas de edad gestacional, hospitalización en periodo neonatal, concentración de TIR (ng/ml), cifra de conductabilidad de ClNa (mEq/l) en el test del sudor y genotipo.
Las mutaciones estudiadas fueron: R347H, R347P, 2789+5G>A, 3120+1G>A, 711+1G>T, R334W, I507del, F508del, 3849+10KbC>T, 1677delTA, 1078delT, V520F, L206W, W1282X, R560T, 2347delG, Q890X, R553X, G551D, S549R(T>G), S549N, M1101K, G542X, 3905insT, Y1092X(C>A), S1251N, 444delA, 1811+1.6kbA>G, 1717-1G>A, R117H, R117C, N1303K, Y122X, 394delTT, G85E, R1066C, 1898+1G>A, W846X, 2184delA, D1152H, CFTRdele2.3, P67L, 2143delIT, E60X, 3659delC, 3272-26A>G, 621+1G>T, A455E, R1162X, R1158X. También analiza la cola poliT del intrón 8 (5T, 7T, 9T) y TGn adyacente.
En segundo lugar, se definieron distintos subgrupos atendiendo a la edad gestacional, considerando RN muy pretérmino (RNMPT) a los inferiores a 32 semanas, pretérmino tardío (RNPT) de 32 a 37 semanas y a término (RNT) de 37 semanas en adelante; y al resultado del cribado neonatal, diferenciando entre sano, portador sano, CFSPID o FQ, según se muestra en tabla 1.
Clasificación por subgrupos de neonatos
| Grupos de recién nacidos | |
|---|---|
| 1. Sano | Cero mutaciones y test del sudor normal |
| 2. Portador sano | Una mutación y test del sudor normal |
| 3. CFSPID | Dos mutaciones, solo una de ellas clasificada como causante de FQ*, con test del sudor normalCero o una mutación y test del sudor dudoso. |
| 4. FQ | Dos mutaciones causantes de FQ y/o test del sudor alterado |
CFSPID: Cystic Fibrosis Screen Positive, Inconclusive Diagnosis; FQ: fibrosis quística.
El nivel de significación estadística fue del 5% y los resultados se analizaron con el paquete estadístico IBM SPSS Statistics Version 15.0.
ResultadosDesde enero de 2011 hasta septiembre de 2019 se detectaron 790RN con cribado neonatal positivo para FQ.
La distribución según la edad gestacional fue de 682RNT (86,3%), 43RNPT (5,4%) y 25RNMPT (3,2%) con edad gestacional inferior a 32 semanas, con un rango entre 24 y 42 semanas de edad gestacional. El 53% de la muestra fueron niñas y el peso de RN medio fue 3.083g, con límites entre 400 y 4.840g. Ingresaron en periodo neonatal el 11,8%.
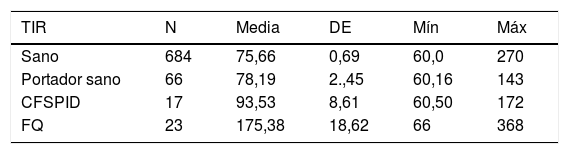
El valor medio de TIR fue 79,16ng/ml (límites 60-367). La concentración media de TIR según subgrupos de pacientes se expone en la tabla 2, siendo en sanos de 75,66ng/ml, para portadores sanos 78,19ng/ml, CFSPID de 93,53ng/ml y para FQ 175,82ng/ml. Se encontraron concentraciones significativamente más elevadas de TIR en afectos de FQ (p<0,001) con respecto a CFSPID, portadores sanos y sanos (tabla 3 y fig. 1).
Media, DE de los valores de TIR (ng/ml) y valores mínimo y máximo según grupos de pacientes
| TIR | N | Media | DE | Mín | Máx |
|---|---|---|---|---|---|
| Sano | 684 | 75,66 | 0,69 | 60,0 | 270 |
| Portador sano | 66 | 78,19 | 2.,45 | 60,16 | 143 |
| CFSPID | 17 | 93,53 | 8,61 | 60,50 | 172 |
| FQ | 23 | 175,38 | 18,62 | 66 | 368 |
CFSPID: Cystic Fibrosis Screen Positive, Inconclusive Diagnosis; FQ: fibrosis quística; TIR: tripsina inmunorreactiva.
Análisis de TIR comparativo según subgrupos de pacientes
| Grupo FQ | Grupo 1 | Grupo 2 | Grupo 3 |
|---|---|---|---|
| Grupo 1 (sano) | |||
| Grupo 2 (portador sano) | p=0,624 | ||
| Grupo 3 (CFSPID) | p=0,027 | p=0,102 | |
| Grupo 4 (FQ) | p<0,001 | p<0,001 | p<0,001 |
En negrita se muestran las diferencias entre grupos estadísticamente significativas.
CFSPID: Cystic Fibrosis Screen Positive, Inconclusive Diagnosis; FQ: fibrosis quística; TIR: tripsina inmunorreactiva.
 atendiendo a la clasificación por subgrupos de pacientes. CFSPID: Cystic Fibrosis Screen Positive, Inconclusive Diagnosis; FQ: fibrosis quística; TIR: tripsina inmunorreactiva.")
También existen niveles superiores de TIR atendiendo al antecedente de prematuridad (p=0,007). Se observaron niveles superiores de TIR en el grupo de RNMPT (mediana 78,35 ng/ml) frente a RNPT (mediana 73ng/ml) o RNT (70,12 ng/ml), siendo esta diferencia estadísticamente significativa.
Se encontraron diferencias significativas en las concentraciones de TIR en el grupo de pacientes ingresados en periodo neonatal (n=93) frente al de no ingresados (n=586), p=0,002. En el grupo de ingresados se obtuvo un valor medio de TIR de 84,50ng/ml (IC 95%: 78,23-90,77) y en el de no ingresados 78,15ng/ml (IC 95%: 75,69-80,62). Esta diferencia es mayor en pacientes ingresados en UCI.
No se observan diferencias en los niveles de TIR en cuanto a sexo o estacionalidad.
El valor medio de la conductabilidad de ClNa en sudor fue 30,56mmol/l (límites 13-270mmol/l). Existe una correlación directa del 64% (p=0,001) entre los niveles de TIR y el test de sudor en pacientes afectos de FQ y CFSPID.
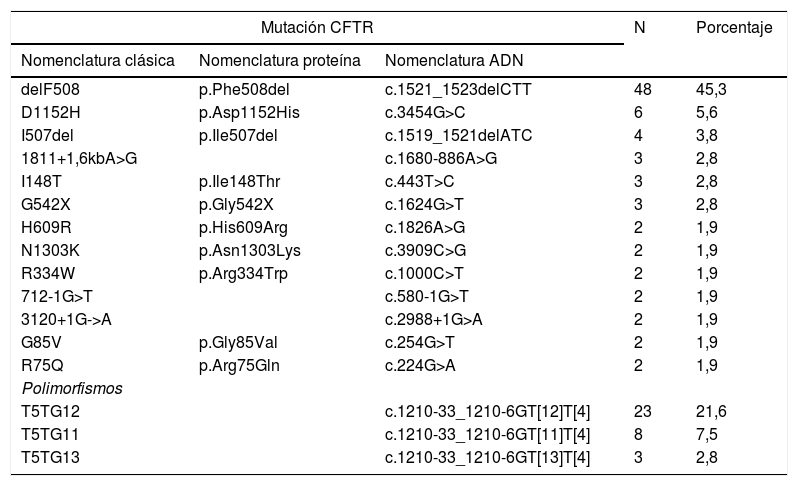
En relación con el estudio genético 684 pacientes (86,6%) no presentaban ninguna mutación en CFTR. La mutación más frecuente fue la denominada clásicamente como delF508, encontrada en forma heterocigota en 40 pacientes (5,06%) y en homocigosis en 8 de ellos (0,9%). En la tabla 4 se muestran las mutaciones y polimorfismos más frecuentes con respecto al total de pacientes con estudio genético alterado (se incluyen portadores sanos, CFSPID y FQ).
Prevalencia de las mutaciones en CFTR más comunes en uno o 2 alelos (n=160)
| Mutación CFTR | N | Porcentaje | ||
|---|---|---|---|---|
| Nomenclatura clásica | Nomenclatura proteína | Nomenclatura ADN | ||
| delF508 | p.Phe508del | c.1521_1523delCTT | 48 | 45,3 |
| D1152H | p.Asp1152His | c.3454G>C | 6 | 5,6 |
| I507del | p.Ile507del | c.1519_1521delATC | 4 | 3,8 |
| 1811+1,6kbA>G | c.1680-886A>G | 3 | 2,8 | |
| I148T | p.Ile148Thr | c.443T>C | 3 | 2,8 |
| G542X | p.Gly542X | c.1624G>T | 3 | 2,8 |
| H609R | p.His609Arg | c.1826A>G | 2 | 1,9 |
| N1303K | p.Asn1303Lys | c.3909C>G | 2 | 1,9 |
| R334W | p.Arg334Trp | c.1000C>T | 2 | 1,9 |
| 712-1G>T | c.580-1G>T | 2 | 1,9 | |
| 3120+1G->A | c.2988+1G>A | 2 | 1,9 | |
| G85V | p.Gly85Val | c.254G>T | 2 | 1,9 |
| R75Q | p.Arg75Gln | c.224G>A | 2 | 1,9 |
| Polimorfismos | ||||
| T5TG12 | c.1210-33_1210-6GT[12]T[4] | 23 | 21,6 | |
| T5TG11 | c.1210-33_1210-6GT[11]T[4] | 8 | 7,5 | |
| T5TG13 | c.1210-33_1210-6GT[13]T[4] | 3 | 2,8 | |
En el número y porcentaje de pacientes se incluyen los portadores de cada mutación en uno o ambos alelos. No se incluyen las mutaciones encontradas en porcentaje inferior al 1%.
CFTR: proteína transportadora de la fibrosis quística; n: número de casos.
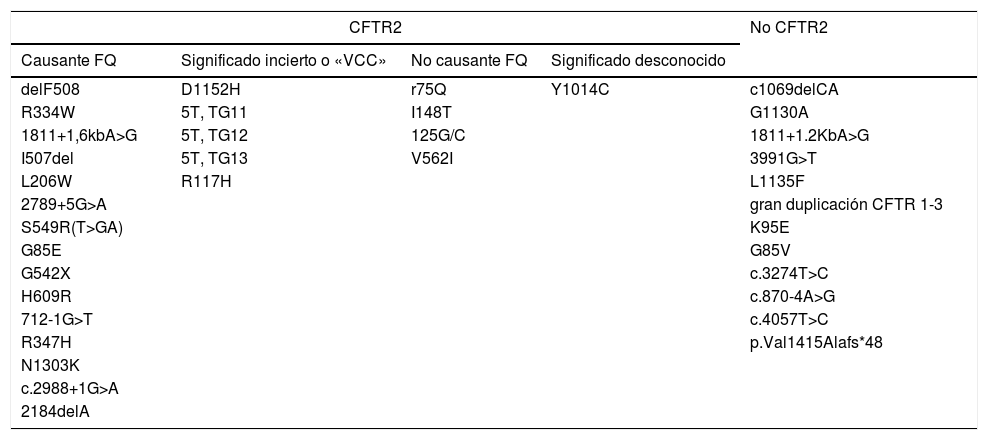
Se categorizan las mutaciones encontradas conforme a la definición contemplada en la base de datos CFTR2 (tabla 5)14. En el subgrupo de afectos de FQ y CFSPID, se observaron concentraciones de TIR significativamente superiores en aquellos portadores de 2 mutaciones causantes de FQ (n=16), valor medio de TIR 190,86ng/ml, con respecto a portadores de una mutación causante y una mutación no causante de FQ (n=4), valor medio 70,22ng/ml (p=0,003) o portadores de una mutación causante y una de significado incierto (n=7), valor medio 108,24ng/ml (p=0,027).
Clasificación de mutaciones encontradas según las recogidas en la base de datos CFTR2
| CFTR2 | No CFTR2 | |||
|---|---|---|---|---|
| Causante FQ | Significado incierto o «VCC» | No causante FQ | Significado desconocido | |
| delF508 | D1152H | r75Q | Y1014C | c1069delCA |
| R334W | 5T, TG11 | I148T | G1130A | |
| 1811+1,6kbA>G | 5T, TG12 | 125G/C | 1811+1.2KbA>G | |
| I507del | 5T, TG13 | V562I | 3991G>T | |
| L206W | R117H | L1135F | ||
| 2789+5G>A | gran duplicación CFTR 1-3 | |||
| S549R(T>GA) | K95E | |||
| G85E | G85V | |||
| G542X | c.3274T>C | |||
| H609R | c.870-4A>G | |||
| 712-1G>T | c.4057T>C | |||
| R347H | p.Val1415Alafs*48 | |||
| N1303K | ||||
| c.2988+1G>A | ||||
| 2184delA | ||||
CFTR2: The Clinical and Functional Translation of CFTR, disponible en http://cftr2.org; VCC: del inglés Varying Clinical Consequence.
Con el fin de optimizar el punto de corte se realiza la curva ROC con un área bajo la curva de 0,93 (p<0,001). Mediante curva ROC se calculó valor de corte de TIR para FQ siendo de 76,2ng/ml, con una sensibilidad del 95,7% y especificidad del 64,5%.
DiscusiónLa reciente introducción del cribado neonatal de FQ mediante la determinación de TIR ha supuesto una mejora evidente en la calidad de vida y supervivencia de estos pacientes al permitir un diagnóstico precoz de la enfermedad15. Sin embargo, se trata de una prueba de cribado y, como tal, presenta una baja especificidad, lo que se traduce en un elevado número de falsos positivos.
El principal inconveniente derivado de su baja especificidad es la ansiedad producida en las familias y el elevado coste que supone la realización del estudio genético y el test del sudor a todo RN con cribado positivo. Por otra parte, ha permitido el diagnóstico de portadores sanos, de individuos afectos de FQ en su forma no clásica e incluso de individuos con mutaciones de significado incierto16,17, cuya inclusión como parte del mismo se encuentra en la actualidad en debate debido al menor beneficio derivado del diagnóstico y mayor estrés provocado en las familias.
En relación con lo anteriormente expuesto se deduce la importancia de la determinación del punto de corte en la concentración de TIR establecido en cada hospital.
Se obtuvo un valor medio de TIR de 79,16ng/ml y un valor máximo de 367ng/ml en una paciente afecta de FQ portadora de 2 mutaciones graves.
En el caso del test de conductabilidad de ClNa en sudor, la gran mayoría de determinaciones realizadas (96,1%) fueron normales, es decir, por debajo del punto de corte de 50 establecido, siendo el valor medio de 30,56mmol/l.
En el análisis por subgrupos se encontraron concentraciones de TIR significativamente superiores en el grupo de afectos de FQ (p<0,001) en comparación tanto con RN sanos y portadores sanos, como los CFSPID. Se encontraron diferencias entre CFSPID y sanos (p=0,027). Estos resultados son consistentes y similares a los presentados en otro estudio llevado a cabo por Parachinni et al., en el cual concluyen que valores más elevados de TIR en la prueba del talón se relacionan con mayor probabilidad con estar afecto de FQ18. Del mismo modo, y al igual que en nuestro estudio, tampoco encontraron diferencias entre el grupo de sanos y portadores sanos.
Sin embargo, algunos estudios de Castellani et al. encuentran diferencias significativas entre sanos y portadores sanos19. Si bien este último punto no se demuestra en la muestra analizada, sí se observa una tendencia a una mayor concentración de TIR en el subgrupo de portadores frente a sanos. No obstante, la distribución de concentraciones de TIR se solapa ampliamente entre los 4 grupos establecidos y, en especial, entre los 3 primeros (sanos, portadores sanos y CFSPID). En consecuencia, la predicción para clasificar a los pacientes en estos 3 grupos en función de los valores de TIR es baja. Por otra parte, la ausencia de significación también podría deberse al menor tamaño muestral con respecto a los estudios mencionados previamente18,19.
La prematuridad puede ser en sí misma una causa de elevación de TIR como postula Korzeniewsli et al., quienes encuentran valores de TIR significativamente superiores en RN de muy bajo peso (<1.500g) y extremadamente prematuros (menores de 28 semanas de edad gestacional)20. Esto coincide con los resultados obtenidos, ya que se observaron niveles superiores de TIR en el grupo de RNMPT frente al grupo de RNPT (p=0,008) y RNT (p=0002).
El estrés y la asfixia perinatal están descritos como una causa de elevación de TIR21, lo que podría tener relación con las diferencias encontradas según el antecedente de ingreso neonatal. En un estudio de Rock et al. se encontró una puntuación de Apgar al minuto y a los 5 minutos significativamente menor en pacientes con elevación de TIR en comparación con la población general22. En el análisis realizado el valor medio de TIR en RN hospitalizados fue mayor que en aquellos sin este antecedente, y este a su vez superior en pacientes ingresados en una unidad de cuidados intensivos neonatales, si bien esto último podría estar en relación con que la mayoría de los pacientes ingresados en UCIN son grandes prematuros.
De acuerdo con lo descrito por otros autores9, un mayor nivel de TIR se correlaciona directamente con una mayor cifra en el test del sudor, siendo esta correlación mayor en pacientes afectos de FQ y CFSPID (r=0,64, p=0,001), lo que implica una correlación lineal del 64% entre ambas variables.
Entre los pacientes portadores de alguna mutación delF508 fue la detectada con mayor frecuencia (45%). Cabe destacar una elevada heterogeneidad molecular en la muestra estudiada, ya que ninguna otra mutación presentó una frecuencia superior al 5% (tabla 4).
Los pacientes portadores de 2 mutaciones causantes de FQ presentaron valores de TIR significativamente superiores. Estos resultados son similares a los registrados en otras series23,24, que muestran concentraciones de TIR y de ClNa en sudor más bajas en pacientes portadores de una mutación no causante de FQ o una mutación rara al compararlos, respectivamente, con portadores de 2 mutaciones causantes de FQ.
Un valor de corte óptimo en nuestro medio para el diagnóstico de FQ sería de 76,2ng/ml, con una sensibilidad del 95,7% y especificidad del 64,5%, lo que supone que, en nuestra población, una concentración plasmática de TIR superior a este valor podría corresponder con mayor probabilidad a una FQ.
En cualquier caso, creemos razonable no elevar el punto de corte en los niveles de TIR de detección del cribado, ya que portadores y pacientes con formas no clásicas quedarían sin diagnosticar, y emplearlo únicamente como un marcador para el diagnóstico precoz. Sería especialmente útil en pacientes con una única mutación identificada en la primera genética en la que esté pendiente el estudio ampliado, siendo más probable que exista otra mutación en aquellos casos con cifras muy elevadas de TIR.
Una de las limitaciones del estudio es su naturaleza observacional y la recogida retrospectiva de datos. Por contra, la principal fortaleza es un tamaño muestral elevado en un estudio realizado en un único centro hospitalario. Serían necesarios estudios prospectivos multicéntricos que apoyen los resultados obtenidos.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
