



Medullary thyroid carcinoma (MTC) is a feature of type 2 multiple endocrine neoplasias (MEN2). Subtype A (MEN2A) is characterised by MTC, pheochromocytoma (50% of patients, usually bilateral) and occasionally parathyroid hyperplasia or adenoma (20%–30% of patients). It is a dominant autosomal hereditary disease that results from germline mutations in the RET proto-oncogene (10q11.2) in 98% of cases.1
We present the case of a girl aged 4 years and 10 months referred after detection of a Cys634Arg mutation in exon 11 of the RET proto-oncogene. The mother had received a diagnosis of MEN2A (with MTC, bilateral suprarenal pheochromocytoma and hyperparathyroidism) five years before; genetic testing was performed in family members, and was positive in our patient. The maternal uncle had died at age 27 years of thyroid carcinoma, and the maternal grandmother had died during childbirth (at age 38 years) from an unknown cause. The relevant medical history of the patient consisted in her use of hearing aids to manage sensorineural hearing loss secondary to a perinatal hypoxic–ischaemic episode.
The patient underwent prophylactic total thyroidectomy at age 4 years and 11 months. During the preoperative period, thyroid function was normal, tests for antithyroglobulin and antimicrosomal antibodies were negative, levels of calcium, phosphorus, alkaline phosphatase and parathyroid hormone (PTH) were normal, and the baseline measurement of calcitonin level was high (16.4pg/ml; normal range, 1–4.8). Urine levels of catecholamines and metanephrines and serum levels of thyroglobulin and carcinoembryonic antigen (CEA) were normal. The findings of the abdominal and thyroid ultrasound examinations were normal. Histopathological examination of thyroid tissue revealed multiple sites of medullary microcarcinoma. Immunochemical assays of neoplastic samples were positive for calcitonin, chromogranin and CEA. The Ki-67 proliferation index was less than 1%, and indicated a benign course.
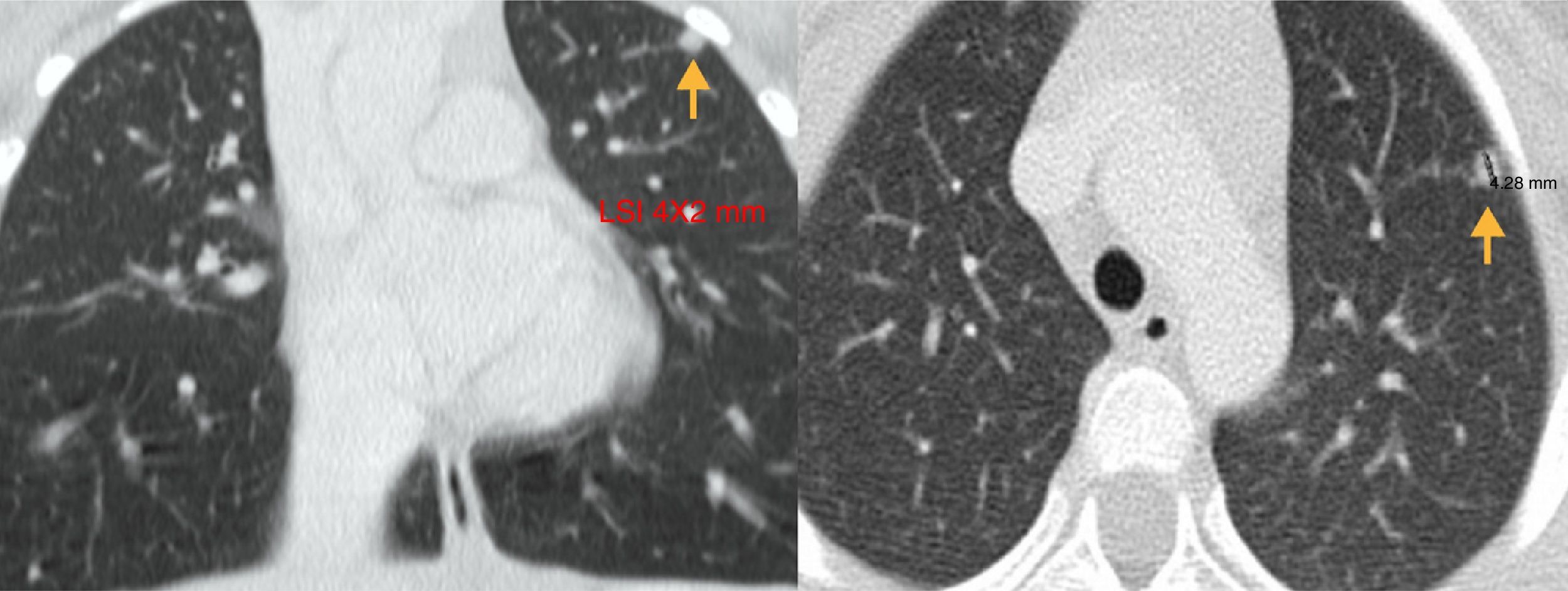
During the postoperative period, the patient had no disturbances of calcium-phosphate metabolism and her calcitonin levels normalised (1.4pg/ml; normal range, 1–4.8), and hormone replacement therapy was initiated with L-thyroxin. The CT scan of the chest and abdomen performed after surgery as part of the staging workup detected a 4×2mm nodule in the upper lobe of the left lung (Fig. 1). Such a finding in the context of MTC associated with a RET mutation, considered to carry high risk, led to suspecting a metastatic lesion as a the most likely possibility, and the patient underwent resection of the lesion guided by CT. The histopathological examination of the lesion revealed that it was a necrotising epithelioid granuloma. This led to the diagnosis of pulmonary tuberculosis, confirmed by a positive Mantoux test (20mm) and testing of gastric lavage and coughed-up sputum specimens. Treatment with isoniazid, pyrazinamide and rifampicin was initiated. In addition, the father was identified as the index case with active pulmonary tuberculosis, and was also prescribed an antituberculosis regimen.
At the time of this writing, the patient remains asymptomatic and receives hormone replacement therapy with levothyroxine at a dose of 150μg/day. Serum levels of calcitonin, metanephrines and CEA are normal.
The presence of an index case of MTC calls for genetic testing of family members. Prophylactic thyroidectomy is the only curative treatment, and the decision to operate is based on the type of mutation (RET genotype),1,2 as there is a 100% correlation between the presence of disease and mutation carrier status.3
The Cys634Arg mutation in exon 11 of the RET proto-oncogene found in our patient and her mother is considered high-risk, and is an indication for total thyroidectomy as soon as possible after diagnosis, preferably before age 4 years.2 Prior to surgery, patients must undergo an evaluation to rule out the presence of pheochromocytoma (yearly measurement of metanephrines in serum and urine) and hyperparathyroidism (calcium PTH levels), starting at age 10 years and, in carriers of the 634 mutation, from the time of diagnosis.4
The postoperative follow-up involves the periodic measurement of serum calcitonin levels accompanied by high-resolution thyroid ultrasound examination.4 Detectable calcitonin levels indicate the presence of distant metastases, and require performance of CT of the neck, mediastinum, lung and liver, and in case a suspicious nodule is detected, an ultrasound-guided fine needle aspiration biopsy.5 In our patient, a lung nodule was detected and resected under CT guidance, which examination identified as a tuberculous necrotising epithelioid cell granuloma.
When patients develop isolated metastases, these should be resected, as radiotherapy and chemotherapy are used as palliative measures. Tyrosine-kinase inhibitors6 have been approved by the FDA for the treatment of metastatic MTC in adults, but have not been authorised for use in the paediatric population.
Please cite this article as: Villamayor Martín R, Bartucci A, Muñoz Calvo MT, Pozo Román J, Argente J. Mutación en gen RET: tiroidectomía profiláctica y valoración postoperatoria. An Pediatr (Barc). 2016;85:266–267.
