
Koolen-de Vries syndrome (KDVS, OMIM 610443) is a rare genetic disorder with an approximate prevalence of 1 per 16,000 live births, with no predominance of either sex. It is characterised by neonatal hypotonia, intellectual disability, dysmorphic features (high and broad forehead, long face, palpebral fissures with epicanthic folds, pear-shaped nose) that become attenuated over the years, and friendly behaviour. Central nervous system abnormalities are also found in 80% of cases, such as epilepsy (50%) and brain malformations (hydrocephalus and agenesis or dysgenesis of the corpus callosum), congenital heart defects (valvulopathies and septal defects) in 40–63% depending on the series, and urogenital anomalies (cryptorchidism, hypospadias, vesicoureteral reflux, hydronephrosis, etc.) in up to 70%.1
It was first described in 2006 as a recurrent microdeletion localised in chromosome 17q21.31, between 500 and 650kb in length, that comprises up to five genes: CRHR1, SPPL2C, MAPT, STH and KANSL1 (or KIAA1267), in addition to two putative genes, MGC57346 and C17orf69. In most cases, these are de novo mutations. Although the role of these genes in the pathogenesis of this disease remains unclear, recent studies have demonstrated that both KANSL1 haploinsufficiency or point mutations are sufficient to produce the disease.2–4 The most recently published case series (with 45 patients) described that while the phenotype is not significantly different based on whether there is a 17q21.31 microdeletion or a nonsense mutation of the KANSL1 gene, there is wide phenotypic variability between individuals.4 A previously described predisposing factor is the inversion of chromosome 17 in one of the parents, which, while necessary, is not sufficient to produce the microdeletion, as it is a very common polymorphism that is found in up to 20% of the European population.1 Only two cases of sibling recurrence have been reported in two independent families in which the mothers had a mosaicism for the chromosome 17 deletion, which could be a risk factor for recurrence, underscoring the importance of offering genetic counselling to parents of affected children.5
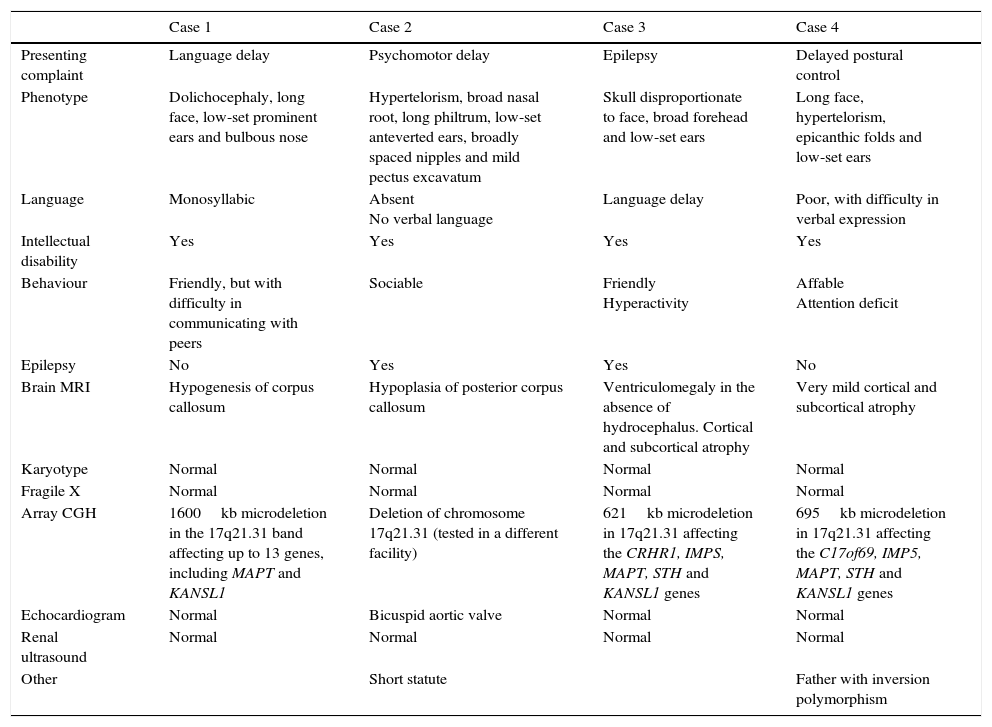
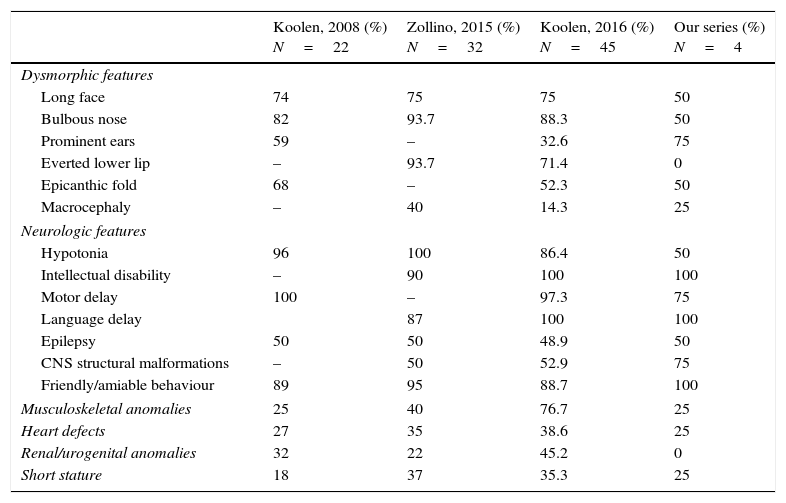
We present a series of four patients with KDVS (Table 1). All patients had some of the clinical features included in the 37 symptoms described by Koolen in the first published series; however, the diagnosis was made when expanded molecular testing was requested following an initial battery of diagnostic tests that were inconclusive. We present a table that summarises what we consider to be the most relevant clinical features described in the most recently published and broader series and those found in our patients (Table 2). Intellectual disability and facial dysmorphism are the most frequently observed features, as was the case in our patients, although potential comorbidities need to be ruled out. Contrary to what has been reported in the literature, none of our patients presented with nephrourologic abnormalities.
Case characteristics.
| Case 1 | Case 2 | Case 3 | Case 4 | |
|---|---|---|---|---|
| Presenting complaint | Language delay | Psychomotor delay | Epilepsy | Delayed postural control |
| Phenotype | Dolichocephaly, long face, low-set prominent ears and bulbous nose | Hypertelorism, broad nasal root, long philtrum, low-set anteverted ears, broadly spaced nipples and mild pectus excavatum | Skull disproportionate to face, broad forehead and low-set ears | Long face, hypertelorism, epicanthic folds and low-set ears |
| Language | Monosyllabic | Absent No verbal language | Language delay | Poor, with difficulty in verbal expression |
| Intellectual disability | Yes | Yes | Yes | Yes |
| Behaviour | Friendly, but with difficulty in communicating with peers | Sociable | Friendly Hyperactivity | Affable Attention deficit |
| Epilepsy | No | Yes | Yes | No |
| Brain MRI | Hypogenesis of corpus callosum | Hypoplasia of posterior corpus callosum | Ventriculomegaly in the absence of hydrocephalus. Cortical and subcortical atrophy | Very mild cortical and subcortical atrophy |
| Karyotype | Normal | Normal | Normal | Normal |
| Fragile X | Normal | Normal | Normal | Normal |
| Array CGH | 1600kb microdeletion in the 17q21.31 band affecting up to 13 genes, including MAPT and KANSL1 | Deletion of chromosome 17q21.31 (tested in a different facility) | 621kb microdeletion in 17q21.31 affecting the CRHR1, IMPS, MAPT, STH and KANSL1 genes | 695kb microdeletion in 17q21.31 affecting the C17of69, IMP5, MAPT, STH and KANSL1 genes |
| Echocardiogram | Normal | Bicuspid aortic valve | Normal | Normal |
| Renal ultrasound | Normal | Normal | Normal | Normal |
| Other | Short statute | Father with inversion polymorphism |
Comparison of the most relevant characteristics found in the most recently published series and in our patients.
| Koolen, 2008 (%) N=22 | Zollino, 2015 (%) N=32 | Koolen, 2016 (%) N=45 | Our series (%) N=4 | |
|---|---|---|---|---|
| Dysmorphic features | ||||
| Long face | 74 | 75 | 75 | 50 |
| Bulbous nose | 82 | 93.7 | 88.3 | 50 |
| Prominent ears | 59 | – | 32.6 | 75 |
| Everted lower lip | – | 93.7 | 71.4 | 0 |
| Epicanthic fold | 68 | – | 52.3 | 50 |
| Macrocephaly | – | 40 | 14.3 | 25 |
| Neurologic features | ||||
| Hypotonia | 96 | 100 | 86.4 | 50 |
| Intellectual disability | – | 90 | 100 | 100 |
| Motor delay | 100 | – | 97.3 | 75 |
| Language delay | 87 | 100 | 100 | |
| Epilepsy | 50 | 50 | 48.9 | 50 |
| CNS structural malformations | – | 50 | 52.9 | 75 |
| Friendly/amiable behaviour | 89 | 95 | 88.7 | 100 |
| Musculoskeletal anomalies | 25 | 40 | 76.7 | 25 |
| Heart defects | 27 | 35 | 38.6 | 25 |
| Renal/urogenital anomalies | 32 | 22 | 45.2 | 0 |
| Short stature | 18 | 37 | 35.3 | 25 |
Genetic testing in all cases identified mutations in one of the five genes that cause the disease. We ought to highlight the size of the mutation in case 1, which exceeded sizes reported to date.
Recently, the first case has been published of KDVS diagnosed prenatally through the detection of bilateral ventriculomegaly in the pregnancy ultrasound examination performed at 33 weeks’ gestation, with confirmation of the microdeletion by means of array-based comparative genomic hybridisation in a sample obtained by amniocentesis.6 This reinforces the importance of detecting the syndrome early, as it not only allows its aetiological diagnosis, but also genetic counselling for the family. Furthermore, considering that this is a fundamentally monogenic disorder, it is likely that it can benefit from pharmacologic treatment targeting the genome or proteome in the future if research is conducted on this field.
In conclusion, KDVS is a rare disease that must be included in the differential diagnosis of patients with unexplained intellectual disability with or without associated dysmorphic features or malformations. Considering the wide clinical variability that has been observed, we believe that genetic hybridisation techniques must be included at the level of brain MRI in the diagnostic evaluation of these patients.
Please cite this article as: Moreno Samos M, Moreno Medinilla EE, Martínez Antón JL, Urda Cardona A. Síndrome Koolen de Vries: un reto en la práctica clínica. An Pediatr (Barc). 2017;86:162–164.
