
El síndrome Koolen de Vries (SKDV, OMIM 610443) es una enfermedad genética infrecuente con una prevalencia aproximada de 1 de cada 16.000 recién nacidos, sin predominio sexual. Se caracteriza por presentar hipotonía al nacimiento, discapacidad intelectual, rasgos dismórficos (frente amplia y ancha, con cara alargada, fisuras palpebrales con pliegues epicánticos, nariz en forma de «pera») que se atenúan con el paso de los años y un comportamiento afable. Además asocian alteraciones en el sistema nervioso central en un 80% de los casos, tales como epilepsia (50%) y malformaciones cerebrales (hidrocefalia y agenesia/disgenesia del cuerpo calloso), defectos cardíacos (valvulopatías y defectos septales) en un 40-63% según las series y anomalías urogenitales (criptorquidia, hipospadia, reflujo vesico-ureteral, hidronefrosis…) hasta un 70%1.
Descrito por primera vez en 2006 como una microdeleción recurrente localizada en el cromosoma 17q21.31, con un tamaño entre 500-650 Kb que abarca hasta 5 genes: CRHR1, SPPL2C, MAPT, STH y KANSL1 (o KIAA1267), además de 2 genes putativos, MGC57346 y C17orf69. Generalmente, son mutaciones de novo. Aunque el papel que desempeñan estos genes en la patogénesis de la enfermedad aún es desconocido, estudios recientes han demostrado que tanto la deleción única de KANSL1 como mutaciones puntuales en este gen son suficientes para producir la enfermedad2-4. En la serie más recientemente publicada (45 pacientes), se describe que aunque el fenotipo no es significativamente distinto si hay una microdeleción de 17q21.31 o una variante truncante del gen KANSL1; al mismo tiempo, existe una gran heterogeneidad fenotípica de un sujeto a otro4. Como factor predisponente se ha descrito la inversión del cromosoma 17 en alguno de los 2 progenitores, aunque si bien es una alteración necesaria no es suficiente para generar la microdeleción ya que es un polimorfismo muy común, encontrándose hasta en el 20% de la población europea1. Tan solo se han documentado 2 casos en 2 familias independientes con hermanos afectados cuyas madres presentaban un mosaicismo en el cromosoma 17, lo cual podría ser un factor de riesgo de recurrencia, de ahí la importancia de proponer consejo genético a los padres con hijos afectados5.
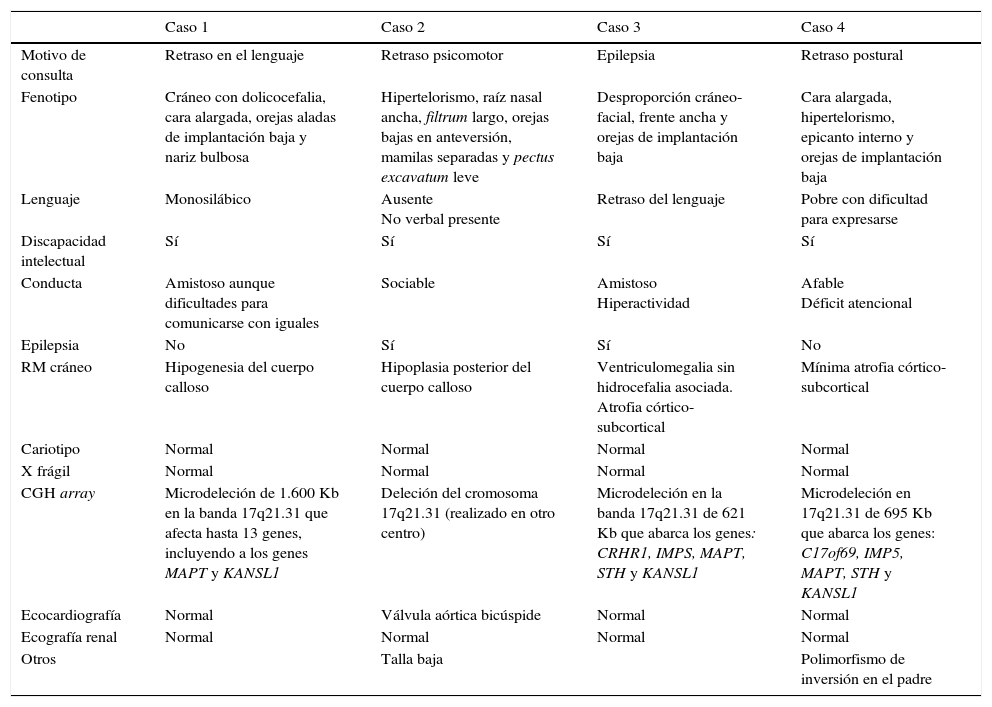
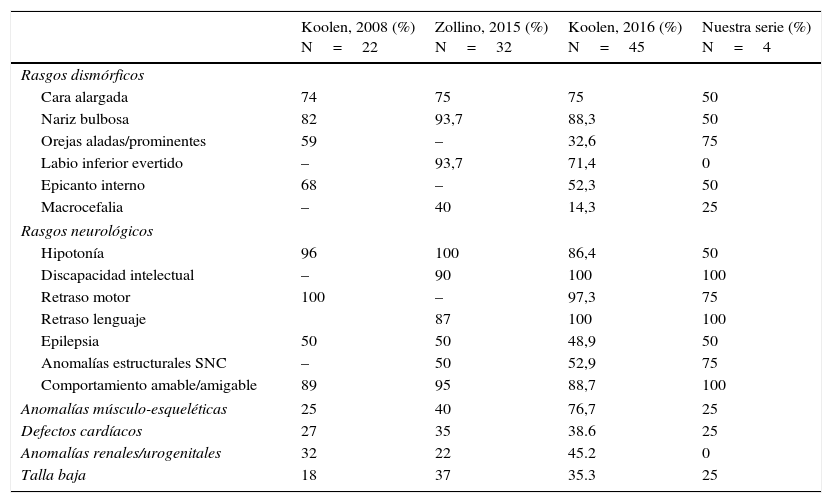
Presentamos una serie de 4 pacientes con SKDV (tabla 1). Todos presentaron algunas de las características clínicas incluidas entre los 37 síntomas descritos por Koolen en su primera serie; sin embargo, el diagnóstico se obtuvo tras una primera batería de pruebas complementarias inconcluyentes, siendo en el seguimiento posterior solicitada la ampliación del estudio molecular. Presentamos una tabla resumen con las características clínicas que consideramos más relevantes descritas en las series más recientes y extensas publicadas, así como las que presentan nuestros pacientes (tabla 2). La discapacidad intelectual, así como el dismorfismo facial, son los rasgos más frecuentemente encontrados, tal y como ocurre en nuestra serie de casos, aunque es necesario descartar comorbilidades asociadas. Pese a lo reportado en la literatura, ninguno de nuestros casos presentó alteraciones nefrourológicas.
Descripción de los casos
| Caso 1 | Caso 2 | Caso 3 | Caso 4 | |
|---|---|---|---|---|
| Motivo de consulta | Retraso en el lenguaje | Retraso psicomotor | Epilepsia | Retraso postural |
| Fenotipo | Cráneo con dolicocefalia, cara alargada, orejas aladas de implantación baja y nariz bulbosa | Hipertelorismo, raíz nasal ancha, filtrum largo, orejas bajas en anteversión, mamilas separadas y pectus excavatum leve | Desproporción cráneo-facial, frente ancha y orejas de implantación baja | Cara alargada, hipertelorismo, epicanto interno y orejas de implantación baja |
| Lenguaje | Monosilábico | Ausente No verbal presente | Retraso del lenguaje | Pobre con dificultad para expresarse |
| Discapacidad intelectual | Sí | Sí | Sí | Sí |
| Conducta | Amistoso aunque dificultades para comunicarse con iguales | Sociable | Amistoso Hiperactividad | Afable Déficit atencional |
| Epilepsia | No | Sí | Sí | No |
| RM cráneo | Hipogenesia del cuerpo calloso | Hipoplasia posterior del cuerpo calloso | Ventriculomegalia sin hidrocefalia asociada. Atrofia córtico-subcortical | Mínima atrofia córtico-subcortical |
| Cariotipo | Normal | Normal | Normal | Normal |
| X frágil | Normal | Normal | Normal | Normal |
| CGH array | Microdeleción de 1.600 Kb en la banda 17q21.31 que afecta hasta 13 genes, incluyendo a los genes MAPT y KANSL1 | Deleción del cromosoma 17q21.31 (realizado en otro centro) | Microdeleción en la banda 17q21.31 de 621 Kb que abarca los genes: CRHR1, IMPS, MAPT, STH y KANSL1 | Microdeleción en 17q21.31 de 695 Kb que abarca los genes: C17of69, IMP5, MAPT, STH y KANSL1 |
| Ecocardiografía | Normal | Válvula aórtica bicúspide | Normal | Normal |
| Ecografía renal | Normal | Normal | Normal | Normal |
| Otros | Talla baja | Polimorfismo de inversión en el padre |
Comparación de las características más relevantes de las últimas series publicadas y nuestros pacientes
| Koolen, 2008 (%) N=22 | Zollino, 2015 (%) N=32 | Koolen, 2016 (%) N=45 | Nuestra serie (%) N=4 | |
|---|---|---|---|---|
| Rasgos dismórficos | ||||
| Cara alargada | 74 | 75 | 75 | 50 |
| Nariz bulbosa | 82 | 93,7 | 88,3 | 50 |
| Orejas aladas/prominentes | 59 | – | 32,6 | 75 |
| Labio inferior evertido | – | 93,7 | 71,4 | 0 |
| Epicanto interno | 68 | – | 52,3 | 50 |
| Macrocefalia | – | 40 | 14,3 | 25 |
| Rasgos neurológicos | ||||
| Hipotonía | 96 | 100 | 86,4 | 50 |
| Discapacidad intelectual | – | 90 | 100 | 100 |
| Retraso motor | 100 | – | 97,3 | 75 |
| Retraso lenguaje | 87 | 100 | 100 | |
| Epilepsia | 50 | 50 | 48,9 | 50 |
| Anomalías estructurales SNC | – | 50 | 52,9 | 75 |
| Comportamiento amable/amigable | 89 | 95 | 88,7 | 100 |
| Anomalías músculo-esqueléticas | 25 | 40 | 76,7 | 25 |
| Defectos cardíacos | 27 | 35 | 38.6 | 25 |
| Anomalías renales/urogenitales | 32 | 22 | 45.2 | 0 |
| Talla baja | 18 | 37 | 35.3 | 25 |
En el estudio genético realizado se halló implicado alguno de los 5 genes responsables de la enfermedad. Destaca el tamaño de la mutación del primer caso, ya que excede lo publicado hasta la fecha.
Recientemente se ha publicado el primer caso de diagnóstico prenatal de SKDV al detectar en la ecografía de control de la semana 33 ventriculomegalia bilateral, confirmándose la microdeleción mediante CGH-array a través de la amniocentesis6. Esto refuerza la importancia de una detección precoz del síndrome, ya que no solo permite el diagnóstico etiológico, sino también proporcionan consejo genético a las familias. Además, teniendo en cuenta que se trata de una enfermedad fundamentalmente monogénica, es de esperar que pueda beneficiarse de terapias farmacológicas sobre el genoma o sobre el proteoma en un futuro, si se emprenden investigaciones al respecto.
En conclusión, el SKDV es una enfermedad rara que debe ser considerada dentro del diagnóstico diferencial de los pacientes con discapacidad intelectual inexplicada asociada o no a algún tipo de dismorfia o malformación. Dada la gran variabilidad clínica existente, consideramos que las técnicas de hibridación genética deben situarse a la altura de la resonancia magnética craneal en el estudio de este tipo de pacientes.
