



Inflammatory myofibroblastic tumour (IMT) is a rare soft tissue tumour in paediatrics. Its histological diagnosis is arduous. Approximately half of all IMTs have positive immunohistochemistry for ALK (anaplastic lymphoma kinase). The prognosis is usually favourable, although the World Health Organisation gives it an intermediate prognosis (7% risk of dissemination and 15% of recurrence).1 Surgical resection has classically been the only treatment option, with little documented efficacy of traditional chemotherapeutic or radiotherapy approaches. For this reason, the emergence of targeted therapy with ALK inhibitors, such as crizotinib, seems promising.1–5 However, the optimal duration of treatment is not well established, and treatment is generally prolonged if the patient maintains response and has significant adverse effects. In fact, it lasts approximately 2 years on average, which is an issue the scientific community should reconsider, given its side effects (vomiting, diarrhoea, neutropaenia, vision disorders, neuropathy), its high cost and the possible emergence of resistance.
We present the case of a 6-year-old girl who presented with a 2-month history of abdominal pain, fatigue, pyrexia, and weight loss. A magnetic resonance imaging (MRI) scan found a mass. It was located on the right side, within the small bowel mesentery, measured 6 × 4 × 6 cm, with homogeneous signal and restricted diffusion, as well as enhancement after gadolinium. It seemed to impair venous drainage from the caecum and terminal ileum, as there were dilated blood vessels surrounding the caecum and hepatic flexure. No lymphadenopathy or other abnormalities were seen. The morphological features were those of a low-grade spindle cell tumour. The immunostaining was positive for SMA, CD117 (cytoplasmic), ALK (cytoplasmic), desmin (patchy), MNF116 (patchy) and vimentin. The expression of INI-1 was preserved.
The findings were suggestive of an IMTSurgery, usually the preferred treatment for localised IMT, could potentially result in short-gut syndrome, making the patient dependent on lifelong parenteral nutrition; therefore, further treatment options were explored. Given the promising results of targeted therapy compared to classic chemotherapy regimens, crizotinib (200 mg daily) was initiated while awaiting the results of molecular studies.
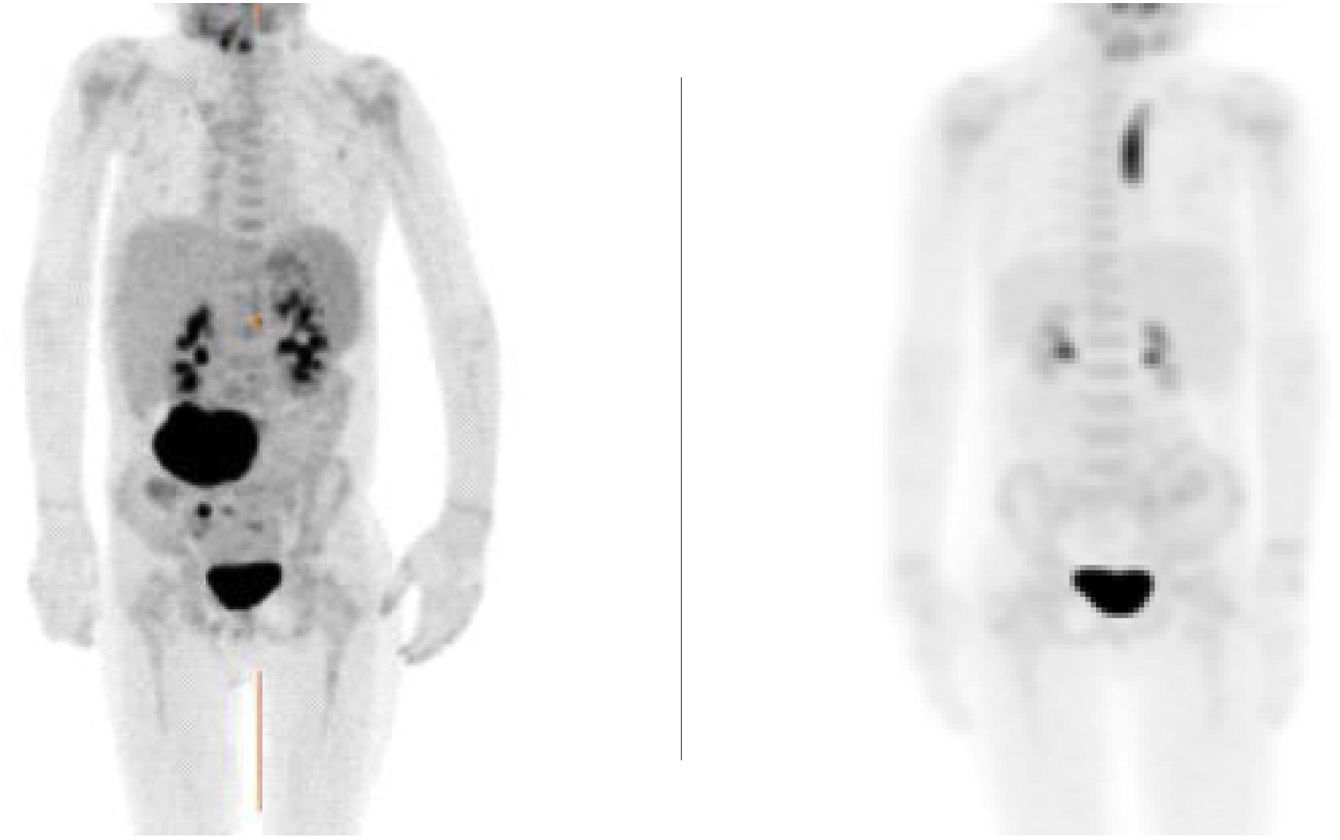
The patient reported progressive improvement of symptoms. Magnetic resonance and positron-emission tomography (PET-CT) scans revealed a small residual lesion at 1 month of treatment and complete resolution of disease at 4 months (Fig. 1).
After 5 months of treatment, the caregivers reported an abnormal running pattern, and the patient started to experience intermittent, unilateral and localised leg pain. An MRI of the abdomen and pelvis was performed, with no evidence of tumour recurrence but with abnormal signal intensity in the upper femora. Blood tests ruled out inflammation and infection. Neuropathy had been previously described as a common side effect (25%) as well as decreased bone formation (uncommon). The symptoms did not improve and crizotinib was thought to be the cause. Treatment was discontinued 1 month later (total duration of treatment of 6 months). The altered gait and joint pain resolved after stopping crizotinib. The patient underwent close follow-up monitoring. At present, it has been 4 years since treatment was completed, and there is no evidence of recurrence and the patient has not required any further treatment.
Whole genome sequencing eventually detected a translocation between ALK and PTRH2/CLTC t(2;17) (p23.2;q23.1).
A review of the literature allows us to appreciate the important role that molecular biology is playing in paediatric patients with IMT. Firstly, it facilitates diagnosis, which is initially erroneous in up to 20% of cases. Pire et al. used fluorescence in situ hybridisation (FISH) or RNA sequencing to detect specific fusions involving ALK, ROS1 or NTRK. These techniques detected tyrosine kinase gene rearrangements in up to 86% of patients with ITM, whereas changes in ALK have been classically reported in approximately 50% of patients.2
In terms of prognosis, recent publications postulate that among the ALK + IMTs, there could be some subtypes with a poor prognosis depending on the detected genetic rearrangements. For example, tumours with RANBP2-ALK rearrangements would not respond adequately to crizotinib and may require treatment with second-generation ALK inhibitors.3
Regarding treatment, recent studies conclude that aggressive surgery is not associated with a better prognosis.1 In inoperable cases, there is controversy about first-line treatment. Some patients have responded to low-dose chemotherapy, but a standard chemotherapy regimen has yet to be established, and there is ample evidence on its side effects. Moreover, the European Paediatric Soft Tissue Sarcoma Study Group and The Children’s Oncology Group have recently published promising results of targeted therapy with ALK-inhibitors.4,5
After the case of our patient, who only received treatment for 6 months with an excellent response, and taking into consideration the side effects of ALK-inhibitors,4,5 we challenge the necessity to continue treatment once complete remission is achieved, regardless of the time it takes to achieve it. In this respect, the question of the long-term efficacy of these treatments also stands out. Our patient has been out of treatment for 4 years, with no signs of recurrence. Therefore, it seems evident that prospective studies with larger cohorts are urgently needed to ascertain the safest way of treating these children. Future trials will need to establish the duration of treatment and the safety of stopping treatment once a complete response has been achieved.
Conflicts of interestThe authors have no conflicts of interest to disclose.
