


Kaposiform haemangioendothelioma (KHE) is a rare, vascular tumour that has little metastatic potential but can be locally aggressive and may be life-threatening. It is diagnosed based on the findings of imaging tests and gross and histological features. It usually presents as a single lesion in the retroperitoneum or subcutaneous tissue, and in 30% of cases it is associated with Kasabach–Merritt syndrome (KMS), a coagulopathy that has complications that are more severe than those of KHE alone. The Sociedad Española de Hematología y Oncología Pediátrica (Spanish Society of Paediatric Haematology and Oncology) proposes surgical resection as first-line treatment, but the involvement of vital structures may preclude this option. In such cases, the use of a combination of antiplatelet therapy and systemic corticosteroid therapy is recommended for first-line treatment, reserving treatment with vincristine, cyclophosphamide, interferon alfa or other cytostatic agents for refractory cases.1
We present the 3 cases of KHE managed in the past decade in our tertiary care hospital, one of whom was a patient with underlying sickle cell disease, a comorbidity that has not been described previously.
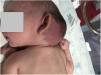
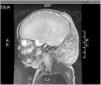
Case 1: full-term infant with a congenital violaceous cervical mass, hard to de touch and extending from the left retroauricular area to the edge of the right preauricular area (Fig. 1). A magnetic resonance image (MRI) scan confirmed the diagnosis of KHE (Fig. 2). The patient presented with thrombocytopenia (25000/dL) and d-dimer elevation (3000μg/L), which suggested the additional presence of KMS. Treatment started with methylprednisolone, antiplatelet therapy and vincristine, which resulted in transient improvement. This was followed by a drop in the platelet count to 5000/dL in the context of necrotizing enterocolitis. Cyclophosphamide was added to the treatment, followed by sirolimus due to nonresponse, which achieved a gradual decrease in tumour size and normalization of the laboratory test results. The patient underwent surgical resection of the mass at age 13 months. At age 22 months, the patient continues to be healthy and without recurrence.
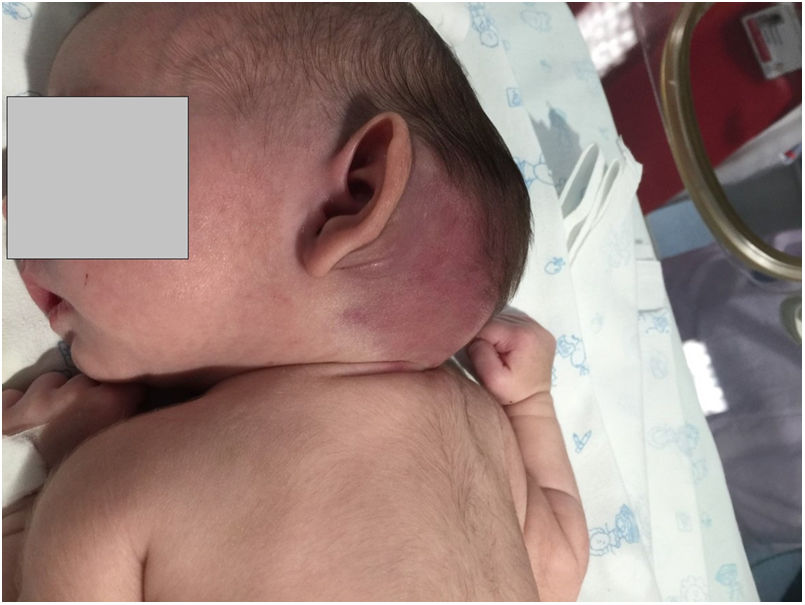
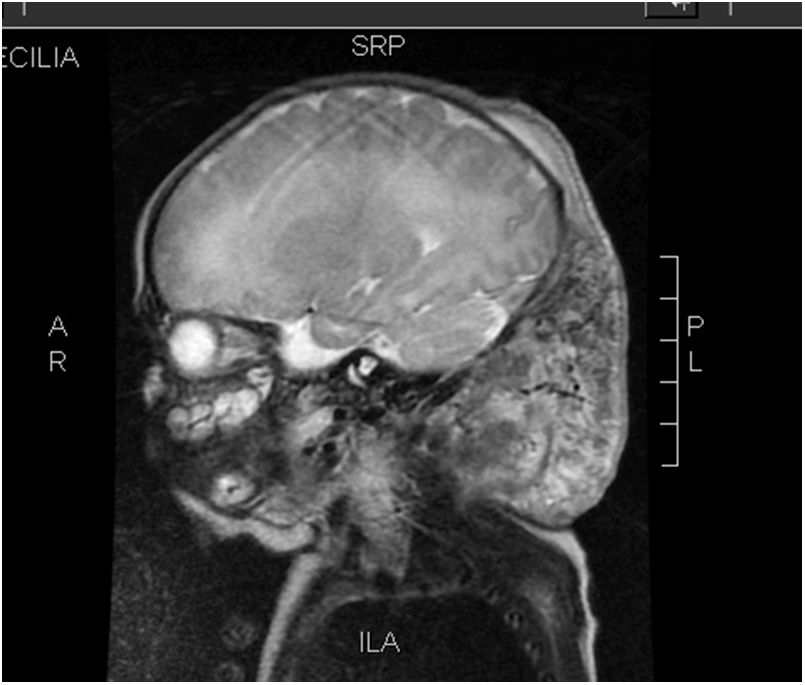
Case 2: girl aged 2 years brought for consultation after detection of an abdominal mass in the course of a febrile illness. The findings of the ultrasound examination, MRI scan and biopsy confirmed the diagnosis of KHE restricted to the muscles of the right abdominal wall, with no laboratory abnormalities suggestive of KMS at onset. Treatment started with methylprednisolone, but it was switched to vincristine after 3 weeks due to nonresponse. After the fifth dose, there was evidence of thrombocytopenia (3000/dL) and d-dimer elevation (12400μg/L) compatible with KMS. This was followed by intraarterial embolization of the tumour by interventional radiology, with little improvement. Since the patient showed little progress, vincristine was discontinued and replaced by everolimus, which achieved a reduction of the tumour. The patient underwent resection of the tumour at age 4 years. At present, the patient is 6 years old and remains disease-free.
Case 3: infant aged 7 months with sickle cell disease presenting with a hard lump in the deep planes of the left arm. After 3 months of follow-up during which a vaso-occlusive crisis had been suspected, the mass increased in size, leading to performance of a biopsy and a confirmed diagnosis of KHE. Although the initial laboratory test results had been normal, at this point the patient exhibited sudden thrombocytopenia (26000/dL) and d-dimer elevation (4430μg/L), suggestive of KMS. Treatment was initiated with vincristine, which achieved a reduction of the tumour size and normalization of the laboratory test results. After the seventh dose, the patient experienced sudden deterioration and was brought to the emergency department with cardiac arrest. The patient exhibited marked splenomegaly, which suggested splenic sequestration secondary to the underlying sickle cell disease. After advanced cardiopulmonary resuscitation failed, the patient was declared dead.
The cases presented here illustrate the considerable heterogeneity found in the management of this disease. Furthermore, given the low incidence of KHE, the literature on the subject is limited to a few case series. This evinces a need for updated guidelines including new therapeutic alternatives. The use of corticosteroids has not been assessed in clinical trials, and most published studies have reported a partial response or a lack of effectiveness when used as monotherapy. Selective tumour embolization seems to achieve a transient improvement in tumour size and platelet counts, so it may be indicated in severe cases as an adjuvant to medical treatment. Total surgical resection, when feasible, is usually the most effective treatment. In some cases, vincristine has achieved complete resolution of the tumour and normalization of laboratory tests.2 Propranolol also appears to be useful in the management of KHE, although the results on this subject are contradictory.3,4 Clinical trials and case studies are currently underway to investigate the use of sirolimus and other mTOR inhibitors, so far with promising results,5,6 and some hospitals are using these drugs as first-line treatment. Although it is a challenging task on account of the low incidence of KHE, there is a clear need for comparative studies of currently used and newly proposed treatments for the purpose of updating management guidelines.
Please cite this article as: Huerta-Calpe S, Pinilla-González A, Juan Ribelles A, Gómez-Chacón Villalba J, Pérez-Tarazona S. ¿Cómo tratamos el hemangioendotelioma kaposiforme? An Pediatr (Barc). 2019;91:122–124.
