
Autosomal recessive polycystic kidney disease (ARPKD) is very important in paediatrics on account of its severity and the early morbidity and mortality it produces. There is considerable variability in its phenotypic expression, most patients are de novo cases, and the exact relationship between genotype and phenotype has yet to be established.1 Due to all of the above, this disease causes a high level of anxiety in families concerning the child's prognosis, as well as a high demand for genetic counselling.
The aim of our study was to provide a retrospective description of a cohort of paediatric patients with ARPKD managed in a single hospital over a 25-year period. We also sought to identify associations between different clinical manifestations and long-term outcomes.
We made a retrospective search to identify patients with a diagnosis of ARPKD followed up in the department of paediatric nephrology between January 1991 and December 2016. The sample included 16 patients (12 male and 4 female) with a median age of 16.5 years at the time of the study. In this sample, 62% of cases were diagnosed before birth based on sonographic findings. The most frequent sonographic feature found in our sample was renal enlargement (75% of cases), followed by the presence of visible renal cysts (72%).
Two patients underwent genetic testing, which identified a R1804fs mutation in gene PKHD1 in one and a double heterozygous mutation in the PKHD1 gene in the other (NM_138694.3: c.3350_3351delTA NP_619639: p.I1117Kfs*7/NM_138694.3: c.3765_3766delinsG NP_619639: p.Q1256Rfs*47).
Two patients died in the neonatal period due to respiratory failure secondary to pulmonary hypoplasia. The 14 remaining patients survive to date. Of these patients, 57% have developed chronic kidney disease (CKD). The median age of this subgroup at the time of this writing is 22.9 years (Table 1).
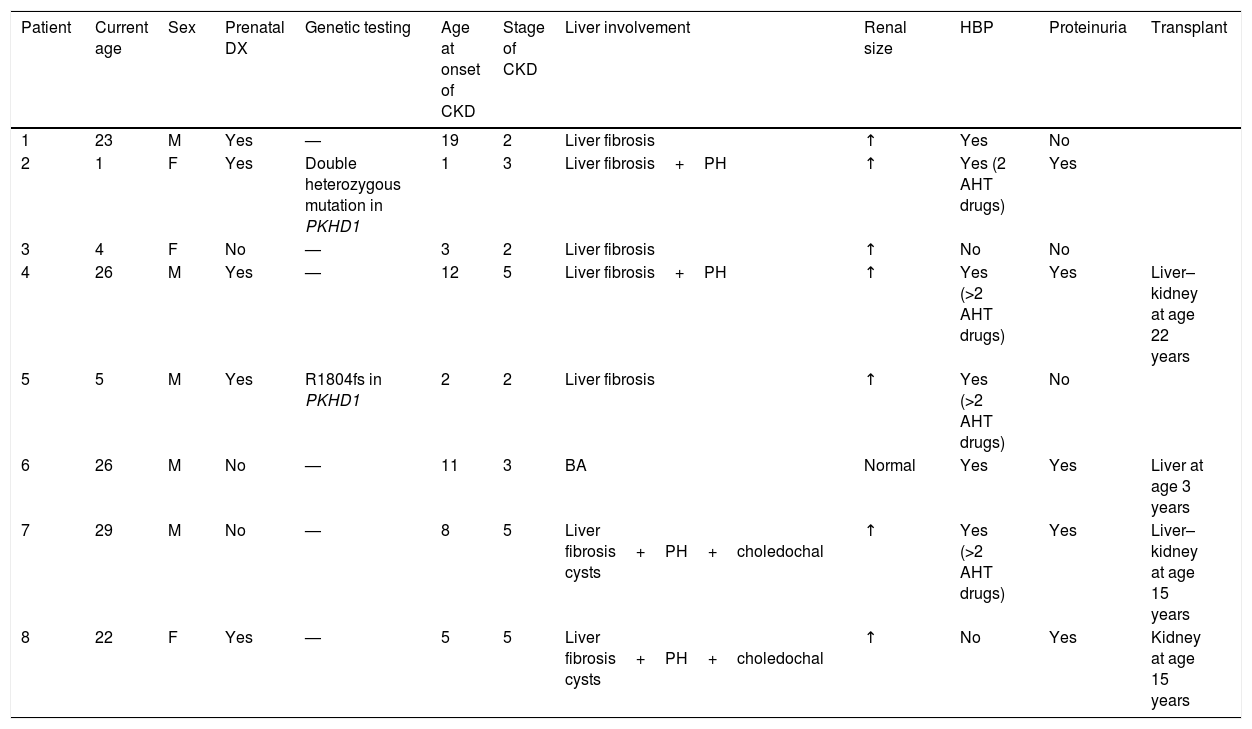
Summary of the data collected for the main variables under study in the group of patients with CKD.
| Patient | Current age | Sex | Prenatal DX | Genetic testing | Age at onset of CKD | Stage of CKD | Liver involvement | Renal size | HBP | Proteinuria | Transplant |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 23 | M | Yes | — | 19 | 2 | Liver fibrosis | ↑ | Yes | No | |
| 2 | 1 | F | Yes | Double heterozygous mutation in PKHD1 | 1 | 3 | Liver fibrosis+PH | ↑ | Yes (2 AHT drugs) | Yes | |
| 3 | 4 | F | No | — | 3 | 2 | Liver fibrosis | ↑ | No | No | |
| 4 | 26 | M | Yes | — | 12 | 5 | Liver fibrosis+PH | ↑ | Yes (>2 AHT drugs) | Yes | Liver–kidney at age 22 years |
| 5 | 5 | M | Yes | R1804fs in PKHD1 | 2 | 2 | Liver fibrosis | ↑ | Yes (>2 AHT drugs) | No | |
| 6 | 26 | M | No | — | 11 | 3 | BA | Normal | Yes | Yes | Liver at age 3 years |
| 7 | 29 | M | No | — | 8 | 5 | Liver fibrosis+PH+choledochal cysts | ↑ | Yes (>2 AHT drugs) | Yes | Liver–kidney at age 15 years |
| 8 | 22 | F | Yes | — | 5 | 5 | Liver fibrosis+PH+choledochal cysts | ↑ | No | Yes | Kidney at age 15 years |
HBP, high blood pressure; AHT drugs, antihypertensive drugs; BA, biliary atresia; CKD, chronic kidney disease; DX, diagnosis; F, female; M, male; PH, portal hypertension.
Current age: age in years at the time of the study; age at onset of CKD: age in years at the time of onset of CKD.
We ought to highlight the rapid progression of disease in the patient with a molecular diagnosis of a double heterozygous mutation, which was consistent with previous studies2 that have described more severe phenotypes in association with the presence of 2 truncating mutations in the PKHD1 gene.
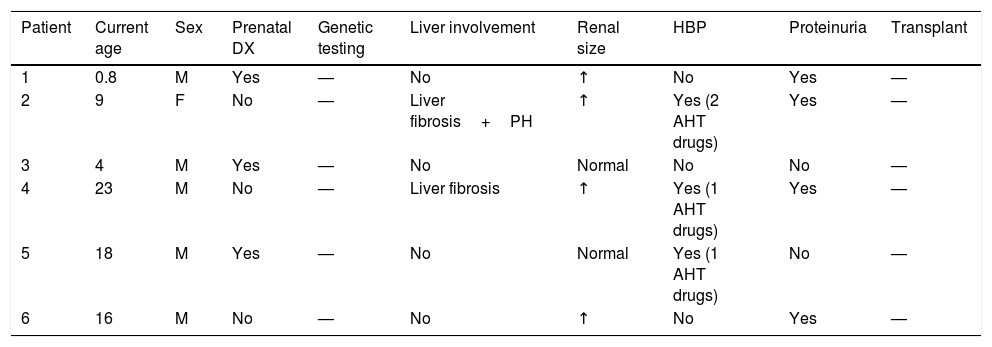
On the other hand, 43% of the sample has yet to develop CKD, and the median age of this subgroup is 12.3 years (Table 2).
Summary of the data collected for the main variables under study in the group of patients without CKD.
| Patient | Current age | Sex | Prenatal DX | Genetic testing | Liver involvement | Renal size | HBP | Proteinuria | Transplant |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.8 | M | Yes | — | No | ↑ | No | Yes | — |
| 2 | 9 | F | No | — | Liver fibrosis+PH | ↑ | Yes (2 AHT drugs) | Yes | — |
| 3 | 4 | M | Yes | — | No | Normal | No | No | — |
| 4 | 23 | M | No | — | Liver fibrosis | ↑ | Yes (1 AHT drugs) | Yes | — |
| 5 | 18 | M | Yes | — | No | Normal | Yes (1 AHT drugs) | No | — |
| 6 | 16 | M | No | — | No | ↑ | No | Yes | — |
HBP, high blood pressure; AHT drugs, antihypertensive drugs; CKD, chronic kidney disease; DX, diagnosis; F, female; M, male; PH, portal hypertension.
Current age: age in years at the time of the study.
We did not find an association between renal size and liver involvement or high blood pressure (HBP) and/or proteinuria. We also found no association between prenatal diagnosis and the development of CKD.
We did find an association between kidney enlargement at the time of diagnosis and future development of CKD (OR, 3.5; 95% CI, 0.24–51.9), although it was not statistically significant (Fisher exact test, P=.53).
In our study, mortality in patients with ARPKD was associated with neonatal pulmonary hypoplasia. Survival was high after the neonatal period. This is consistent with previous reports in the literature,3 although previous authors reported a higher mortality compared to the one found in our sample. This improvement in survival may be due to previous studies4 being a few years older and not reflecting the impact of recent advances in neonatal care and of the follow-up of patients in specialised referral units. Prenatal diagnosis, which occurs in most cases at present, and the option of terminating the pregnancy constitute another factor that needs to be taken into account when it comes to the epidemiology of ARPKD.
The morbidity found in survivors of ARPKD is due to HBP that is difficult to control and the development of CKD and liver disease, whereas lung function develops correctly. In agreement with the literature, more than 50% of the patients progressed to CKD. In previously published case series,5 kidney impairment developed in the first decade of life, but in our sample 43% of the patients had normal renal function past age 10 years.
Several authors6 have suggested that renal size could be a parameter indicative of severity of disease. In our sample, we found an association between kidney enlargement and the development of CKD, although it was not statistically significant, a fact that could be due to the small sample size.
Our study shows that ARPKD is a chronic disease that is treatable and with long-term patient survival past the neonatal period, manifesting with clinically significant HBP of early onset and, in rare cases, with severe proteinuria.
Chief among the limitations of our study is the small sample size, which precludes the extrapolation of our findings to the larger population and reduced the statistical power of the analyses. In addition, only 2 patients underwent genetic testing, which limited our ability to draw conclusions regarding the role of genetic testing in the management of this disease.
Please cite this article as: Rubio San Simón A, Carbayo Jiménez T, Vara Martín J, Alonso Díaz C, Espino Hernández M. Poliquistosis renal autosómica recesiva en el siglo xxi: seguimiento y evolución a largo plazo. An Pediatr (Barc). 2019;91:120–122.
