



Neonatal haemochromatosis (NH) is the leading cause of acute liver failure in the neonatal period.1 Recent discoveries on its aetiology and pathogenesis have led to a radical shift in its management and prognosis.1,2 In 95% of cases, the aetiology involves an alloimmune disease occurring in the gestational period.1,3 The clinical presentation of NH varies widely and is nonspecific. The classic presentation includes hypoglycaemia, jaundice and coagulopathy. The definitive diagnosis is based on the detection of iron accumulation in extrahepatic tissues4 through examination of biopsy samples or magnetic resonance imaging (MRI). Immunochemical evidence of complement-mediated liver injury is characteristic of gestational alloimmune liver disease (GALD).2 Traditional treatment (iron chelators and antioxidants) was associated with a survival of less than 20%, and patients typically required a liver transplant.1 New treatments (exchange transfusion and intravenous immunoglobulin therapy [IVIG]) have increased survival without transplantation to 75%.5 The most important change is that prenatal prevention has now become possible with administration of IVIG during pregnancy, with an efficacy nearing 100%,6 once the diagnosis of GALD is confirmed.
The aim of the review presented here was to analyse the cases of NH managed in our department in a period of 10 years and to assess the impact of recent advances in terms of patient outcomes and survival. We conducted a retrospective study by reviewing the cases of NH diagnosed between January 2005 and December 2014 in our hospital. We collected data on gestational age and birth weight, family history, obstetric history (including findings of prenatal ultrasound examinations), clinical manifestations and timing of onset, manifestations during the course of the disease, laboratory abnormalities, findings of imaging tests, comorbidities, histopathological features, time elapsed to diagnosis, treatment received and patient outcomes.
We identified 6 cases of NH in the past 10 years (4 in girls and 2 in boys). There was no relevant family history in any case. All mothers were healthy, all but one had had previous pregnancies, and 3 had a history of miscarriage. In 4 cases, there was a history of oligohydramnios and in 3 cases a history of intrauterine growth restriction. Three of the patients were born preterm and another 3 were small for gestational age.
There was considerable variation in the clinical presentation. Three patients had onset within 24h from birth with jaundice and exhibited progressive liver injury. Another patient required admission to the neonatal intensive care unit due to extreme prematurity and developed hepatomegaly and cholestasis at 12 days post birth, with subsequent progression of liver injury. Another patient was admitted due to a prenatal diagnosis of congenital heart disease and dysmorphic features, and developed refractory shock of unknown origin, oliguria and liver failure in the first hours of life. The last patient presented with refractory shock and multiple organ failure from birth and had a fulminant progression that led to death 5 days post birth. All patients had kidney failure. Two patients had hypothyroidism, one of them associated with pancreatic insufficiency. All had anaemia, 4 had thrombocytopenia and 1 pancytopenia. There was a moderate transaminase elevation. All patients had coagulopathy, cholestasis, hypoalbuminemia, elevation of serum ferritin and iron and saturation of transferrin and α-fetoprotein. The initial abdominal ultrasound scan was normal in 4 patients, with abnormal findings in subsequent scans. Four patients underwent an MRI examination, which evinced iron overload in the liver and pancreas.
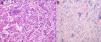
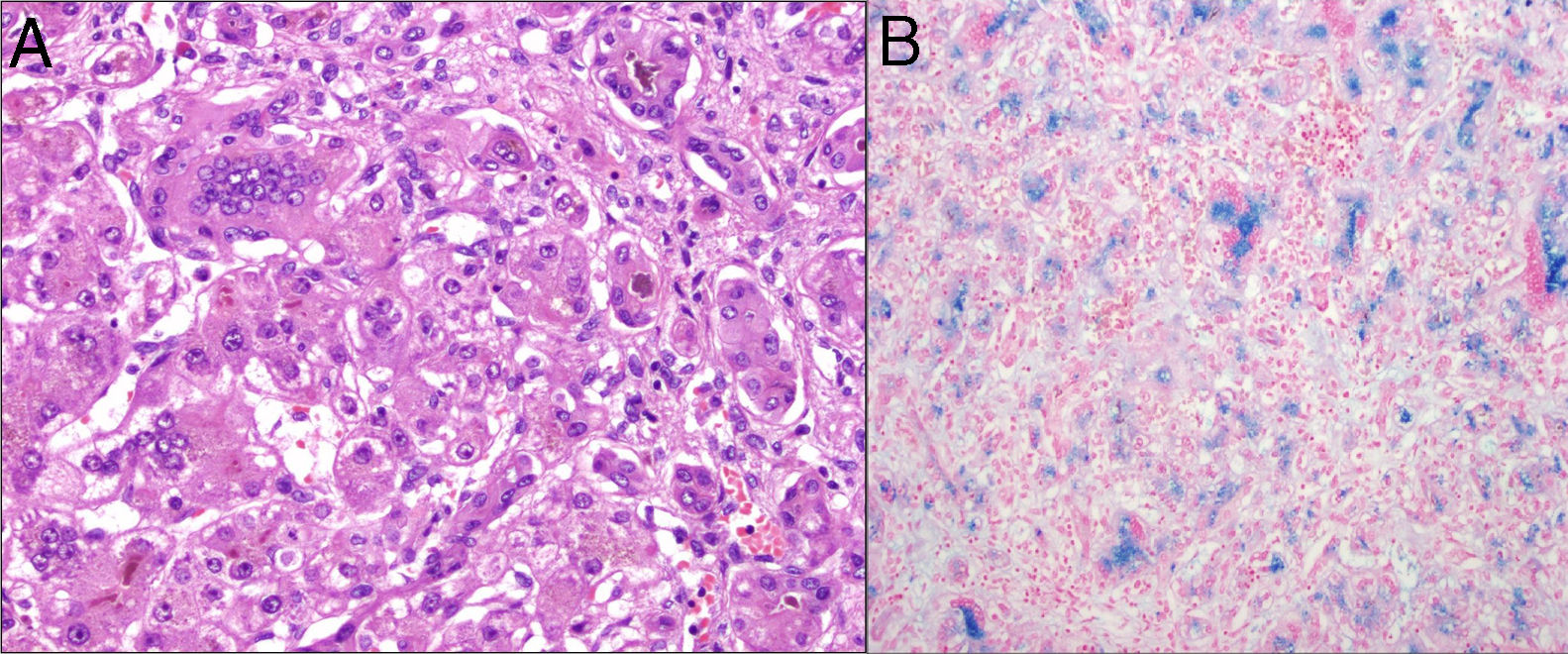
The diagnosis was made by examination of open liver (Fig. 1) and oral mucosa biopsy samples in 5 patients, and during the post-mortem examination in the remaining patient. In one of the patients, the diagnosis was also made based on the positive results of immunohistochemical analysis.
As regards treatment, 3 patients received exchange transfusions and 2 received IVIG. Three were treated with antioxidants and iron chelators. All patients showed a limited response to treatment. Two of the patients treated most recently received a liver transplant, and they are the patients that have survived to present. All other patients died (Table 1). The mother of one of the patients received prenatal preventive treatment in the following pregnancy and gave birth to a healthy child.
Relevant features.
| Patient | GA (weeks) | Weight (g) | Age at onset | Clinical features at onset | Comorbidities | Peak total/direct bilirubin (mg/dL) | PeakAST/ALT(IU/L) | Age at diagnosis (dpb) | Treatment | Transplant | Death (dpb) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 26.5 | 610 | 12dpb | Abdominal distension, hepatomegaly and cholestasis | Hypothyroidism | 11/10.6 | 29/21 | 66 | Supportive care | No | Yes (67) |
| 2 | 39 | 2290 | First 24h | Shock, anasarca | Dysmorphic features and heart disease | 30/20 | 199/63 | 15 | ChelatorsAntioxidants | No | Yes (>134) |
| 3 | 40.5 | 2650 | First 24h | Jaundice | 52/36 | 1445/260 | 21 | ChelatorsAntioxidants | No | Yes (85) | |
| 4 | 37 | 2360 | First 24h | Episodes of hypoglycaemia, hepatomegaly, jaundice, ascites | HypothyroidismPancreatic insufficiency | 25/20 | 229/110 | 22 | EarlyaExchange transfusionIVIG | Yes | No |
| 5 | 34 | 2000 | First 24h | Shock, anasarca | 6/4 | 115/38 | Post mortem | EarlyaExchange transfusion | No | Yes (5) | |
| 6 | 35.1 | 2370 | First 24h | Oedema, jaundice | 37/23 | 100/37 | 13 | EarlyaExchange transfusionIVIGChelatorsAntioxidants | Yes | No |
ALT, alanine aminotransferase; AST, aspartate aminotransferase; dpb, days post birth; GA, gestational age; IVIG, intravenous immunoglobulin therapy.
The findings of our review are mostly consistent with other reports in the literature with a few exceptions. There is no previous description of the comorbidities we found in 2 of the patients (hypothyroidism and pancreatic insufficiency), and we believe they may be of interest, as they could be related to the alloimmune aetiology. Also, survival without transplantation in our patients was not as good as reported by other authors, while the age at diagnosis and the time of treatment initiation were similar to those described in other case series.
To conclude, in the past few years neonatal haemochromatosis has gone from being an untreatable orphan disease to a preventable and curable disease. A high level of suspicion for this diagnosis is key to initiate treatment that has proven effective as early as possible. Diagnosis of this disease is essential for its prevention in future generations.
Please cite this article as: García Victori E, Mañas R, Castilla Fernández Y, Ruiz Campillo CW, Castillo Salinas F. Hemocromatosis neonatal. Diez años en un cambio de paradigma. An Pediatr (Barc). 2019;91:124–126.
