

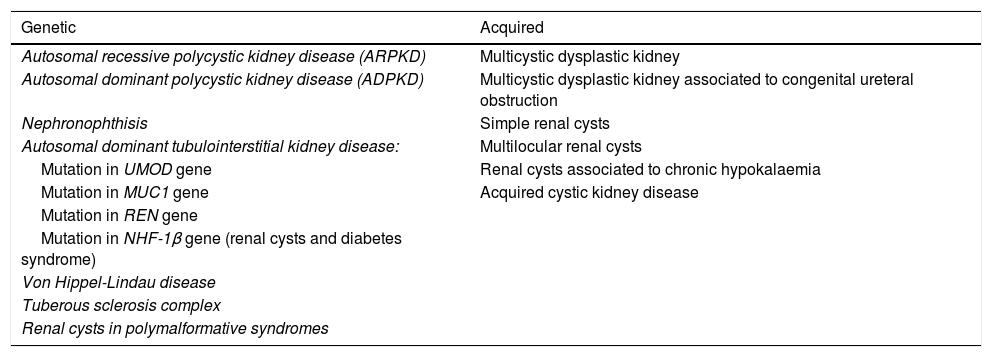
The presence of hyperechoic kidneys during a prenatal ultrasound evaluation can be indicative of a wide variety of renal diseases, ranging from clinically insignificant transient involvement to diseases with progressive deterioration of renal function.1 This hyperechogenicity is frequently a manifestation of renal cystic disease.1,2 There are many conditions that manifest with renal cysts, both genetic and acquired (Table 1),3,4 and they may have onset from intrauterine life through adulthood. The most frequent ones are autosomal dominant polycystic kidney disease, autosomal recessive polycystic kidney disease, nephronophthisis and those recently reclassified as autosomal dominant tubulointerstitial kidney disease.4 The differential diagnosis must take into account the ultrasound findings, the family history and the presence of extrarenal abnormalities.
Diseases associated with renal cysts.
| Genetic | Acquired |
|---|---|
| Autosomal recessive polycystic kidney disease (ARPKD) | Multicystic dysplastic kidney |
| Autosomal dominant polycystic kidney disease (ADPKD) | Multicystic dysplastic kidney associated to congenital ureteral obstruction |
| Nephronophthisis | Simple renal cysts |
| Autosomal dominant tubulointerstitial kidney disease: | Multilocular renal cysts |
| Mutation in UMOD gene | Renal cysts associated to chronic hypokalaemia |
| Mutation in MUC1 gene | Acquired cystic kidney disease |
| Mutation in REN gene | |
| Mutation in NHF-1β gene (renal cysts and diabetes syndrome) | |
| Von Hippel-Lindau disease | |
| Tuberous sclerosis complex | |
| Renal cysts in polymalformative syndromes |
We present the case of a family with renal cystic disease of a genetic aetiology in which the index case that led to diagnosis was a newborn in whom prenatal ultrasound had detected increased echogenicity in both kidneys.
The patient underwent an evaluation at 1-month post birth for assessment of the prenatal bilateral renal hyperechogenicity. The postnatal ultrasound examination confirmed increased echogenicity in the renal cortex with presence of microcysts (Fig. 1).

The personal history was: product of a twin pregnancy achieved by in vitro fertilization due to primary infertility (fallopian tube obstruction). Maternal gestational diabetes treated with insulin. The prenatal ultrasound examination at week 20 revealed increased echogenicity in the kidneys that persisted until birth, with diminished corticomedullary differentiation, normal kidney size and absence of cysts. This was associated with polyhydramnios from week 26. There were signs of preterm labour at 32 weeks that required treatment with tocolytics and steroids to promote lung maturation. The patient was delivered by caesarean section at 37 weeks’ gestation with a birth weight of 2095g. He was admitted to the neonatal ward due to transient feeding intolerance.
The family history was: healthy father, mother with history of spondylodiscitis in L4-L5 treated with a graft, tuberculous pleural effusion at age 20 years and allergic rhinitis. No history of urologic disease, chronic renal disease or renal replacement therapy. Healthy twin sister except for evidence in the postnatal ultrasound of a nonspecific abnormality in the gallbladder.
Both parents underwent an ultrasound examination. The mother had a few clinically insignificant renal cysts at the initial evaluation, but her glomerular filtration rate showed progressive deterioration until reaching chronic kidney disease stage G2 to G3a A1. Magnetic resonance imaging revealed multiple renal cysts with progressive development of ureteropelvic junction stenosis in the left kidney. The mother exhibited a sustained mild liver function impairment with evidence of a liver haemangioma on imaging. The mother had a second pregnancy, this time spontaneous, also with insulin-dependent gestational diabetes, and once again prenatal ultrasound detected increased echogenicity in the kidneys of the foetus starting at week 20, associated with polyhydramnios from week 30. The postnatal ultrasound confirmed the bilateral renal hyperechogenicity with presence of microcysts.
Given the evidence supporting a hereditary genetic cause, including bilateral renal hyperechogenicity in 2 foetuses of a mother with a history fallopian tube obstruction, insulin-dependent gestational diabetes, chronic kidney disease with cysts ureteropelvic junction stenosis and mild liver function impairment, we ordered a genetic evaluation to assess for suspected renal cysts and diabetes syndrome.
This syndrome is caused by mutations in the gene encoding human transcription factor 2, also known as hepatocyte nuclear factor-1β (HNF-1β; 17q12).1,2,5,6 This factor is involved in the organogenesis of various tissues in the pancreas, liver and genitourinary tract, which explains the phenotype of this disorder. It has an autosomal dominant pattern of inheritance. The most common features of this syndrome are renal cysts and maturity-onset diabetes of the young (MODY), with other features of variable frequency including pancreatic, genital, hepatic, metabolic and neurologic abnormalities.5 Mutations in the HNF-1β gene correspond to a broad phenotypic spectrum, and large case series published in the literature have shown a wide range of renal impairment, from foetal death due to prenatal kidney failure to normal renal function in adulthood, the degree of which is not associated with the genotype.1,6 Mutations in the HNF-1β gene are one of the main causes of hyperechoic kidneys in the foetus.1,2
The patient underwent molecular testing for assessment of small deletions, insertions and point mutations in the splice sites and coding regions of the PKD1, PKD2 and HNF-1β genes, which detected the disease-causing mutation c.695delG (p.Arg232Profs*33) in exon 3 of the HNF-1β gene. This was subsequently confirmed by Sanger sequencing. We found that the mutation exhibited cosegregation with the disease that run in the family, as the variant identified in the patient turned out to have a maternal origin (affected mother) while the father was not a carrier (healthy). Carrier testing revealed that the sister (healthy) was not a carrier, while the brother (affected) had the mutation.
During the follow-up, the index patient exhibited reduced renal growth, a glomerular filtration rate of approximately 75mL/min/1.73m2 at 20 months of age, impaired urinary concentrating capacity and hyperparathyroidism and normal urine albumin levels and blood pressure. His younger brother is currently exhibiting a low glomerular filtration rate for age with impaired urinary concentrating capacity.
Please cite this article as: Hernández PR, Molina AG, Pérez AG, Swafiri ST, Carbajo EP. Estudio de hiperecogenicidad renal: del fenotipo al genotipo en la mutación del gen del factor nuclear hepático1β. An Pediatr (Barc). 2019;90:315–316.
