




Malformación en mano y pie hendido en una misma familia con monosomía 22q11 y 17p13.3
B. Gener 1, M. del Campo 1, E. González-Roca 2, M. Salido 3, C. Orejuela 4, O. Villa 3, D. Godal 4, M. Vilella 4, F. Solé 3 y L.A. Pérez-Jurado 1
1Unitat de Genètica. Department de Ciències Experimentals i de la Salut. Universitat Pompeu Fabra. Barcelona. 2Laboratorio de Microarray. Laboratorio Centro de Regulación Genómica. Barcelona. 3Laboratorio de Citogenética. Hospital del Mar. Barcelona. 4Residència Marinada. Grup Pere-Mata. Reus. Tarragona. España.
Presentamos el caso de un paciente varón de 30 años con retraso mental severo, pezones invertidos, hipospadias glandular y anomalías en extremidades consistentes en monodactilia de ambas manos y pie hendido bilateral. El cariotipo estándar mostró 45,XY, der(17)t(17;22)(p13.3;q11.21),-22. Esta anomalía citogenética también se identificó en la hermana, una mujer con retraso mental, hipoplasia mamaria y pie hendido unilaterales, y en la madre, la cual presentaba dificultades de aprendizaje sin anomalías de extremidades asociadas. Asimismo, los tres individuos tienen talla baja, voz nasal y rasgos dismórficos característicos.
El estudio de hibridación in situ con fluorescencia (FISH) realizado en el probando y la madre descartó deleción de LIS1 en 17p13 y confirmó la deleción de la región del síndrome de Di George/síndrome velocardiofacial (DGS/VCFS) en 22q11.2 (sonda TUPLE) en todas las células estudiadas. El estudio con microarray CGH (hibridación genómica comparada) con ensamblaje completo de clones del cromosoma 22 realizados en estos dos pacientes, ha puesto de manifiesto una deleción que se extiende desde el centrómero hasta 1,5 Mb de la región crítica comúnmente delecionada en el DGS/VCFS.
Según nuestro conocimiento, este es el primer caso descrito de ectrodactilia en un paciente con una anomalía cromosómica que afecta a los cromosomas 22 o 17. Si bien es cierto, que se han descrito anomalías leves de extremidades en pacientes con el fenotipo y deleción clásica del DGS/VCFS, ninguno de los 5 loci conocidos asociados a ectrodactilia implican los cromosomas mencionados. Nuestros hallazgos sugieren que la monosomía 22q10-q11.21 y/o 17q13.3 podrían alterar las vías normales de desarrollo de manos, pies, genitales y mama. En el momento actual se están desarrollando estudios dirigidos a la identificación de los posibles genes candidatos responsables de las anomalías descritas.
Presentación de 2 casos con monosomía 1p36
E. Mansilla 1, F. López-Grondona 1, L. Rodríguez 1, M.L. Martínez-Fernández 1, R.M. Arteaga 2, J. Gómez-Ullate 2 y M.L. Martínez-Frías 1,3
1ECEMC. Centro de Investigación sobre Anomalías Congénitas (CIAC). Instituto de Salud Carlos III. Madrid. 2Departamento de Pediatría. Hospital Marqués de Valdecilla. Santander. 3Departamento de Farmacología. Facultad de Medicina. Universidad Complutense. Madrid. España.
Introducción: Con el avance de las técnicas de citogenética cada vez son más los casos descritos con monosomía 1p36, considerada hoy un síndrome de genes contiguos. Esto ha permitido definir un patrón fenotípico que hace posible sospechar dicha anomalía (hipotonía, epilepsia, retraso mental y del crecimiento, y rasgos dismórficos). Se presentan 2 casos con deleción terminal 1p remitidos a nuestro centro por su neuropediatra para estudio citogenético de alta resolución, por presentar epilepsia rebelde al tratamiento y retraso mental severo no filiado.
Casos clínicos:Caso 1. Niña de 3 meses que presenta: peso 4.280 g (P < 3), talla 57 cm (P10) y PC 38,5 cm (P10); ojos profundos, frente prominente, fontanela amplia, hipotonía generalizada, reflejos osteotendinosos disminuidos y nulo sostén cefálico. Entre sus antecedentes destaca la presencia de episodios de crisis tónico-clónicas que responden mal al tratamiento. Caso 2. Varón de 3 años que presenta: peso 13.600 g (P10), talla 96,2 cm (P10-25), PC 43,5 cm (P < 3); occipital y facies aplanados, paladar ojival, hipotonía generalizada, hiporreflexia osteotendinosa, nula sedestación, nulo lenguaje. Al nacimiento se le diagnostica enfermedad de Ebstein y refiere desde el primer mes de vida episodios de espasmos rebeldes al tratamiento.
En los dos se realizó cariotipo de alta resolución, que sugería la existencia de una deleción terminal en el brazo corto del cromosoma 1 que fue confirmada mediante técnicas de hibridación in situ con fluorescencia (FISH) con la sonda específica de la región subtelomérica del cromosoma 1: 46,XX/XY, del(1) (p36.22).ish(tel 1p-/1q +) de novo.
Ambos casos comparten manifestaciones clínicas que concuerdan con las descritas dentro de la monosomía 1p36, que deben plantear la realización de un cariotipo de alta resolución. Si resulta normal, además deberían descartarse reordenamientos subteloméricos.
Agradecimientos: Este trabajo forma parte del proyecto (PI020028) que ha sido financiado por el Fondo de Investigaciones Sanitarias (FIS), Instituto de Salud Carlos III. Ministerio de Sanidad y Consumo.
Trisomía 20q y monosomía xp21-pter derivada de una translocación t(x;20) materna en una niña con dismorfias
M. Santos 1, G. Rodríguez-Criado 2, A. Meneses 2, F. Llanes 2 y C. Fuster 1
1Unitat de Biologia. Facultat de Medicina. Universitat Autònoma de Barcelona. 2Instituto Hispalense de Pediatría. Sevilla. España.
Introducción: En contraste con la trisomía parcial 20p reiteradamente descrita, los individuos con trisomía parcial 20q son raros. La mayoría de los casos son debidos a la presencia en uno de los progenitores de la translocación equilibrada t (?;20).
Objetivo: Identificar mediante técnicas de citogenética molecular el cromosoma derivativo, der(X), presente en el cariotipo de una niña de 30 meses que presentaba dismorfismo cuya madre era portadora de la translocación t(X;20)(p21;q11.2). Al nacer presentaba cuello corto con piel abundante, acortamiento rizomiélico de las extremidades, hiperlaxitud articular y piel redundante en miembros, edema en el dorso de los pies, asimetrías, cierta anquilosis de codos y el estudio cardiológico diagnosticó CIA ostium secundum. A los 9 meses presentaba retraso de crecimiento, ojos mongoloides, hendiduras palpebrales hacia arriba, paladar estrecho y alto, cuello corto y surco simiesco bilateral, surco de ambos pies engrosados. La niña empezó a andar a los 28 meses, siendo su edad ósea retrasada y a los 30 meses presentaba dificultad en el lenguaje.
Resultados: Los perfiles de la hibridación genómica comparada de alta resolución (HR-CGH) evidencian de forma clara la ganancia de 20q y pérdida de Xp21-pter. La aplicación posterior de la hibridación in situ fluorescente (FISH) multicolor de 24 colores ha permitido confirmar la presencia de material del cromosoma 20 adicionado en Xp.
Discusión: Hasta el momento se han descrito 10 pacientes con trisomía parcial 20q. Sus alteraciones fenotípicas están también relacionadas con la pérdida del cromosoma implicado en la translocación; ninguna de las translocaciones descritas involucraba el cromosoma X. El estudio comparativo de los datos clínicos de estos pacientes evidencia la presencia de alteraciones cardíacas, paladar estrecho y alto, cuello corto, dificultad en el habla y retraso en el crecimiento.
Agradecimientos: Financiación recibida por SAF (2003-03894) y CIRIT (2001, SGR-00201). Beca de la Generalitat de Catalunya (2002FI 00281).
De novo der(x)t(x;14)(q24;q13): caracterización y manifestaciones fenotípicas
E. Guillén-Navarro 1, I. López 2, G. Glover 3, J.A. Bafalliu 2 y A. Mínguez 4
Unidades de 1Genética Médica del Servicio de Pediatría, 2Citogenética y 3Genética Molecular del Centro de Bioquímica y Genética Clínica. 4Servicio de Oftalmología Infantil. Hospital Universitario Virgen de la Arrixaca. Murcia. España.
Introducción: La combinación de técnicas citogenéticas y moleculares permiten caracterizar actualmente las alteraciones cromosómicas de forma más precisa. Aún así la predicción del fenotipo en el caso de translocaciones X; autosoma es difícil. Presentamos el caso de una paciente con una translocación desequilibrada X;14.
Caso clínico: Lactante, mujer, de 4 meses y medio de edad remitida a la consulta por fotofobia y rasgos dismórficos. Primera hija de padres jóvenes, sanos y no consanguíneos. Embarazo sin incidencias. Parto a término, eutócico. Período neonatal: APGAR, 9/10; P: 1.980 g; L: 41 cm, y PC: 31 cm. Taquipnea transitoria. Otoemisiones acústicas positivas. Cribado neonatal normal. A la exploración física presenta P: 5.560 g (P < 3); L: 56 cm (P < 3); PC: 40,5 (P25), dismorfia craneofacial (escaso relieve de arcos ciliares, exoftalmos, puente nasal alto, nariz picuda, labios finos, retrognatia y fosita preauricular izquierda), clinodactilia en quinto dedo de la mano izquierda y angioma plano frontal. Sostén cefálico presente, sintonización y conexión defectuosa. Evaluación oftalmológica normal. Ecocardiografía: fosa oval permeable. Ecografía renal normal. TC craneal 3D: cierre de la parte más posterior de las suturas lambdoideas. Cariotipo de alta resolución e hibridación in situ con fluorescencia (FISH): 46,X,der(X),t(X;14)(q24;q13).ishder(x)(wcp14 + ,telXq-). El cariotipo de los padres fue normal. Estudios de replicación tardía usando BudR y moleculares en proceso.
Comentarios: Las manifestaciones clínicas de las translocaciones desequilibradas X;14 van a depender de los puntos de rotura cromosómica y de la extensión de la inactivación del cromosoma 14 en el der(X). Hasta la fecha existen 5 casos publicados de translocación desequilibrada X;14, y sólo dos de ellos han dado lugar a trisomía funcional 14q. Se compara este caso con los descritos en la bibliografía.
Translocación críptica subtelomérica t(2;10)(q37.3;q26.1) segregando en una familia con aparición de dos segregantes recíprocos
M. Palomares 1, A. Delicado 1, P. Lapunzina 1, M.L. de Torres 1, M.A. Mori 1, L. Fernández 1, M.C. Roche 2, J. Arcas 2, M. Orera 3, C. Aritmendi 4 e I. López-Pajares 1
Servicios de 1Genética Médica y 2Neurología Pediátrica. Hospital Universitario La Paz. 3Unidad de Genética y 4Servicio de Pediatría. Hospital General Universitario Gregorio Marañón. Madrid. España.
Caso clínico: Primer hijo de matrimonio sano, no consanguíneo. Antecedentes familiares: una tía paterna fallecida a los 7 meses de vida con malformaciones congénitas. En la exploración al nacimiento se observaron rasgos faciales dismórficos con facies aplanada y redonda, hipoplasia supraorbitaria, hendiduras palpebrales pequeñas y nariz pequeña con narinas antevertidas. En la radiografía de esqueleto se aprecian costillas gráciles y edad ósea avanzada. El fondo de ojo reveló un coloboma corneorretiniano. Por este motivo se solicitó un cariotipo que resultó normal con un nivel de bandas GTL de 550.
A la edad de 13 meses el paciente fue remitido al servicio de Genética Médica por presentar retraso psicomotor y rasgos dismórficos. En la RM craneal se observó una ampliación del cuarto ventrículo, megacisterna, una leve pérdida auditiva bilateral de 20 dB y anomalías esqueléticas en manos y pies.
Estudios de citogenética molecular: El cribado de las regiones subteloméricas mediante técnicas de hibridación in situ con fluorescencia (FISH) reveló un der(2)t(2;10)(q37.3;q26.1) con monosomía 2q terminal asociada a trisomía 10q terminal. La extensión de dicho estudio mostró que el padre y la abuela paterna eran portadores de una translocación en balance entre los brazos largos de los cromosomas 2 y 10. Estudios posteriores de FISH en otros miembros de la familia permitieron identificar un der(10)t(2;10)(q37.3;q26.1) en una niña de 10 meses con trisomía distal 2q asociada a monosomía 10q. Los hallazgos clínicos eran distintos a los del paciente, destacando el retraso psicomotor, los rasgos dismórficos y una craneosinostosis.
Estudios de genética molecular: El tamaño de los fragmentos implicados fue caracterizado a través del análisis marcadores microsatélites en los cromosomas 2q y 10q. Éstos demostraron que las regiones envueltas en la reestructuración se extendían desde el telómero al menos 4,4 Mb en el cromosoma 2 y 6,9 Mb en el cromosoma 10.
Este trabajo resalta la importancia de realizar estudios de las regiones subteloméricas en todos aquellos pacientes con retraso psicomotor de etiología no aclarada, en especial en aquellos casos acompañados de rasgos dismórficos, anomalías congénitas y con una historia familiar de retraso mental.
Agradecimientos: Este trabajo es apoyado por una beca (PI030617) del Fondo de Investigación Sanitaria (FIS) del Instituto de Salud Carlos III. Ministerio de Sanidad y Consumo.
Reestructuraciones subteloméricas en pacientes con retraso mental y/o dismorfia usando MLPA
M.P. Madero, E. Domínguez y M. Tamparillas
Centro de Análisis Genéticos. Zaragoza. España.
Introducción: Las reestructuraciones subteloméricas se han establecido como el factor genético causante de retraso mental en el 5-7 % de los pacientes afectados estudiados. Sin embargo, en la mayoría de los defectos subteloméricos no se han establecido relaciones con fenotipos clínicos determinados. Por esta razón el cribado de todas las regiones subteloméricas es un diagnóstico muy valioso en pacientes que cumplan al menos tres de los requisitos establecidos por De Vries (De Vries et al, 2001): historia familiar de retraso mental, retraso del desarrollo prenatal, anomalías de desarrollo posnatal, dos o más rasgos faciales dismórficos, uno o más rasgos dismórficos no faciales y malformaciones congénitas.
El método más común de identificación de estas alteraciones ha sido la hibridación in situ fluorescente (FISH) pero este es caro y laborioso. Un método alternativo es el MLPA (Multiplex Ligationdependent Probe Amplification) que permite la cuantificación rápida y precisa de todas las regiones subteloméricas.
Pacientes y métodos: Se han analizado 50 pacientes, afectados de retraso mental y/o dismorfia con resultado previo de normalidad en cariotipo (bandas GTG, nivel 550 bandas) y sin síndromes clínicos reconocibles, mediante la técnica MLPA usando un nuevo set de sondas subteloméricas (the SALSA P036 Human Telomere Test Kit. Disponible en: http://www.mrc-holland.com). Las reestructuraciones encontradas fueron validadas y chequeadas por FISH (sondas TEL Qbiogen).
Resultados: Se han detectado reestructuraciones subteloméricas en el 12 % de los pacientes (6/50), siendo estas reorganizaciones las siguientes: 2 deleciones (4p y 19p) y 6 amplificaciones (5q, 16p, 12p, 20p [2x], 21q). En 3 pacientes las reorganizaciones fueron combinadas (deleciones/amplificaciones).
Conclusiones: Este estudio muestra que la técnica MLPA es rápida, fiable y relativamente barata para detección de reestructuraciones subteloméricas. La simplicidad de esta técnica la hace altamente recomendable para el cribado diagnóstico de rutina de cambios en el número de copias de regiones teloméricas submicroscópicas en pacientes con retraso mental, asociado o no a dismorfias.
Mosaicismo en el patrón de inactivación del cromosoma X en una paciente con retraso mental nacida tras FIV-ICSI.
I. Lorda-Sánchez 1, D. Diego-Álvarez 1, E. Silva 2, M.J. Trujillo-Tiebas 1, D. Cantalapiedra 1, C. Ramos 1 y C. Ayuso 1
1Servicio de Genética. Fundación Jiménez Díaz. Madrid. España. 2Departamento de Oftalmología. Universidad de Coimbra. Portugal.
La inactivación de uno de los cromosomas X en mujeres ocurre al azar durante el período embrionario como método de compensación de dosis. La proporción relativa de cada línea celular, en la que bien el cromosoma X paterno (Xp) o el materno (Xm) permanecen inactivos, tiende a ser del 50 %. Este patrón de inactivación puede variar debido a alteraciones estructurales o a mutaciones en algún gen de ese cromosoma. Está demostrado que la inactivación al azar de Xp o Xm ocurre en el blastocisto, pero permanece la controversia sobre el patrón de la inactivación de Xp desde la fecundación hasta este momento.
Presentamos el caso de una paciente nacida tras embarazo gemelar bicorial logrado mediante FIV-ICSI. Al nacimiento presentó bajo peso, hipotonía, ectasia piélica y atresia de coanas. A los 5 años destacaba retraso psicomotor, marcha atáxica, asimetría facial y corporal, y presencia de zonas hiperpigmentadas. La RM mostró asimetría con dilatación ventricular derecha. Hipoplasia de carótida. Cariotipo de alta resolución: 46,XX; prueba de hibridación in situ fluorescente (FISH) para el síndrome de Williams y Angelman: negativa.
El ensayo de metilación en el gen HUMARA (Xq11.2) mostró inactivación cercana al 95 % del alelo materno en sangre periférica. El estudio en biopsia de piel en dos zonas de diferente pigmentación mostró cariotipo normal en ambas y un patrón de inactivación de Xp y Xm no aleatorio (metilación del 90 % de un alelo respecto del otro) y complementario entre ellas.
Este patrón no aleatorio de inactivación refleja una situación de mosaicismo, en el que podría estar implicado un gen ligado al cromosoma X. Dada la alteración en la metilación descrita tras la técnica de FIV-ICSI, la posibilidad de que la técnica haya influido en dicho patrón queda abierta a discusión.
Riesgo para defectos congénitos oculares en niños nacidos tras fecundaciónin vitro
E. Bermejo 1, G. Dequino 2, L. Cuevas 1, J. Mendioroz 1, D. Prieto 3 y M.L. Martínez-Frías 4
Secciones de 1Epidemiología del ECEMC y 2Teratología Clínica y SITTE. Centro de Investigación sobre Anomalías Congénitas (CIAC). Instituto de Salud Carlos III. Ministerio de Sanidad y Consumo. 3Departamento de Ciencias Sanitarias. Universidad de Alcalá de Henares. 4Directora del ECEMC y del CIAC. Instituto de Salud Carlos III. Ministerio de Sanidad y Consumo. Profesora del Departamento de Farmacología de la Facultad de Medicina. Universidad Complutense. Madrid. España.
En 1978 nació el primer "bebé probeta", Louise Brown, en el Reino Unido, y en 1984 nacía el primero en España. Se estima que entre el 13 y el 24 % de las parejas en edad reproductiva (según datos de la Sociedad Española de Fertilidad: http://www. sefertilidad.com/infobasica/infogeneral/breve.php) tienen problemas de fertilidad, y estudios recientes sugieren que esa frecuencia podría verse incrementada en los próximos años por la influencia de multitud de factores. El empleo de las técnicas de reproducción asistida ha hecho factible que muchas parejas infértiles puedan tener hijos. En consecuencia, el número de niños que nacen cada año tras la aplicación de dichas técnicas ha ido creciendo a lo largo de las últimas décadas en todo el mundo. Sin embargo, son ya muchos los estudios que cuestionan la inocuidad de esos procedimientos y, mientras es clara su asociación con gestaciones múltiples y complicaciones relacionadas (fundamentalmente prematuridad), no hay conclusiones tan claras y universales en lo que respecta al riesgo para defectos congénitos.
En este trabajo hemos analizado epidemiológicamente, sobre los datos del ECEMC (Estudio Colaborativo Español de Malformaciones Congénitas), la hipótesis sugerida en 2001 por Anteby et al. Estos autores encontraron una frecuencia elevada de anomalías oculares en una cohorte de 47 niños nacidos tras fecundación in vitro (FIV), aunque discutían las limitaciones de su estudio al tratarse de una muestra sesgada. Posteriormente, Cruysberg et al (2002) y Moll et al (2003), apoyaron tal hipótesis mediante la publicación de 1 y 5 casos, respectivamente, con retinoblastoma.
En los datos del ECEMC hemos observado un incremento significativo del riesgo global para defectos congénitos oculares mayores (detectados durante los 3 primeros días de vida), entre los niños nacidos tras FIV o ICSI (OR: 4,91; IC 95 %: 1,29-13,29; p = 0,00007). Tras aplicar diversos modelos de regresión logística condicionada, condicionando por tiempo y lugar de nacimiento, y controlando la gemelaridad, observamos que, al menos en los embarazos no gemelares, existe un incremento significativo del riesgo, derivado de la FIV, en relación con los defectos mencionados (OR: 4,41; IC 95 %: 1,20-16,24; p = 0,026). No se pudo controlar el efecto de la etnia debido al tamaño de la muestra para las etnias diferentes a la blanca.
Por tanto, podría estar justificado incluir un examen oftalmológico detallado dentro de los protocolos de seguimiento de los niños nacidos tras FIV.
Exposición prenatal a corticoides para maduración del pulmón fetal: efectos sobre el peso, talla y perímetro cefálico del recién nacido dependiendo del número de ciclos administrados
E. Rodríguez-Pinilla 1, G. Dequino 1, C. Mejías 1, B. Rato 1, P. Fernández 1, D. Prieto 2 y M.L. Martínez-Frías 1
1Sección de Teratología Clínica. Centro de Investigación sobre Anomalías Congénitas (CIAC). Instituto de Salud Carlos III. 2Departamento de Ciencias Sanitarias. Universidad de Alcalá de Henares. Madrid. España.
Introducción: El uso prenatal de corticoides (betametasona/dexametasona) para la inducción de la maduración pulmonar fetal es una práctica habitual en medicina perinatal, con el total reconocimiento de sus efectos beneficiosos sobre la morbilidad y mortalidad por el síndrome de dificultad respiratoria (SDR) en el prematuro. En los últimos años han aparecido numerosos trabajos mostrando efectos adversos de la exposición prenatal a estos corticoides, en especial tras la administración de dos o más ciclos (disminución del peso y PC, miocardiopatía hipertrófica transitoria, incremento de la mortalidad, supresión adrenal, sepsis neonatal y leucomalacia).
En este trabajo analizamos en una muestra de más de 30.000 recién nacidos consecutivos (sin defectos congénitos) el impacto sobre el crecimiento fetal tras la exposición a un ciclo o a dos o más en embarazos con amenaza de parto prematuro.
Material y métodos: Hemos utilizado los datos de los RN controles del Estudio Colaborativo Español de Malformaciones Congénitas (ECEMC). Los análisis se han realizado estratificando por edad gestacional. Para el análisis estadístico hemos utilizado la t de Student para comparación de medias y un modelo de Regresión Lineal para control de factores de confusión.
Resultados: La exposición a betametasona/dexametasona durante el sexto o séptimo mes de gestación disminuye significativamente la media del peso (573 g, p < 0,0005), de la talla (2,79 cm, p < 0,0005) y del PC (1,28 cm, p < 0,0005) en el recién nacido. El impacto negativo sobre las tres medidas fue mayor entre los expuestos a dos ciclos o más (comparados con los expuestos a un ciclo), si bien la diferencia alcanzó significación estadística sólo para el PC (disminución media en prematuros tras un ciclo de 0,04 cm, y tras dos ciclos o más de 2,49 cm). El 29 % de las mujeres, habían recibido múltiples ciclos de corticoides.
Conclusiones: No existe justificación para administrar más de un ciclo de corticoides en embarazadas con amenaza de parto prematuro. No obstante, la administración de un solo ciclo conlleva también una significativa disminución del peso y de la talla del recién nacido prematuro.
Heredabilidad de las fisuras labiopalatinas en una muestra de población española
M.D. Saavedra 1, M. Orera 2, J. Fermosel 3 y C. Bañuelos 4
1Fundación Investigación. 2Unidad de Genética. 3Servicio de Pediatría. 4Cirugía Plástica Infantil. Hospital General Universitario Gregorio Marañón. Madrid. España.
Introducción: La heredabilidad de un rasgo multifactorial indica la proporción de la variabilidad del rasgo que es atribuible a factores genéticos, por lo que es una indicación de la posibilidad de recurrencia. Depende del género del afectado, de la magnitud de la afección, del número de familiares involucrados y del grado de parentesco. La fisura labiopalatina aislada y unilateral es más frecuente en varones, mientras que la fisura palatina bilateral es más frecuente en mujeres.
El riesgo de recurrencia varía en las distintas poblaciones según la frecuencia basal poblacional específica, y dentro de cada familia, dependiendo del número de genes de susceptibilidad heredados, que puede inferirse a partir de la morfología facial.
Objetivo: Determinar la heredabilidad de la fisura labiopalatina en una muestra de pacientes de Madrid.
Material y método: Se han analizado 39 pacientes procedentes de la AFILAPA (Asociación de Fisurados de Labio y Paladar) y del Hospital General Universitario Gregorio Marañón, que participaron voluntariamente en el estudio. Se realizó una valoración clínica, descartándose las formas sindrómicas y aquellas en las que pudieron contribuir factores teratógenos. Se examinaron también los progenitores y hermanos de los afectados. Posteriormente se procedió al análisis de los árboles genealógicos. La estimación del riesgo de recurrencia se ajustó al tamaño de la hermandad.
Resultados: El estudio se ve dificultado por el tamaño de la muestra y la procreación limitada de la población española actual. Se han analizado 39 familias de las cuales se han incluido 33 y se han excluido seis. De los 33 pacientes analizados, 21 son hombres y 12 son mujeres. La heredabilidad calculada para el labio hendido (con o sin paladar hendido es 4,7 %). La heredabilidad del paladar hendido aislado es 5,9 %.
Discusión: Se observa una heredabilidad algo mayor para el paladar hendido aislado que para el labio hendido. Las cifras obtenidas son menores que las observadas en poblaciones orientales y caucásicas. Es preciso ampliar el estudio para obtener riesgos más precisos y para poder identificar los genes de susceptibilidad propios de nuestra población.
Agradecimientos: Trabajo financiado por la Red Temática de Investigación Cooperativa, Genética Clínica y Molecular.
Gemelaridad y reproducción asistida en pacientes con síndrome de Beckwith-Wiedemann. Datos del Registro de Síndromes de Sobrecrecimiento
P. Lapunzina, L. Magano, A. Delicado, M.L. de Torres, M.A. Mori, F. Cabañas, M. Segovia, I. Incera, P. Arias, I. López Pajares y R. Gracia
Servicio de Genética, Neonatología, Genética Molecular y Endocrinología Infantil. Hospital Universitario La Paz. Madrid. España.
Varias publicaciones en los últimos años han sugerido con preocupación la posibilidad de un aumento de la incidencia de enfermedades raras que afectan el imprinting genómico en niños nacidos después de técnicas de reproducción asistida (TRA). Se ha focalizado primariamente sobre los potenciales problemas epigenéticos que se producen después del cultivo de embriones, el clonado de núcleos de células somáticas y las TRA.
Por otra parte, los gemelos monozigóticos se observan más frecuentemente en los pacientes con síndrome de Beckwith-Wiedemann (SBW) que en la población general, y muchos de estos gemelos son discordantes para el SBW. Tres estudios diferentes apuntaron a una asociación entre TRA y SBW. Siete pacientes con SBW de Estados Unidos, seis de Francia, siete del Reino Unido y otros cuatro de otros países de Europa han nacido con SBW después de TRA.
El Registro de Síndromes de Sobrecrecimiento (SSC) ha comenzado su actividad en el año 2003 con el objeto de recoger información de los SSC. Todo paciente con SSC es incluido en una base de datos específica, y muchos de éstos tienen asimismo muestras biológicas para diagnóstico y/o investigación. Actualmente cuenta con contribuciones de más de 30 hospitales y centros diferentes.
Sobre 41 pacientes con SBW con datos completos en el Registro, 5 niños presentaban antecedentes de gemelaridad y/o habían nacido mediante TRA.
En la tabla se exponen las características principales de estos pacientes:
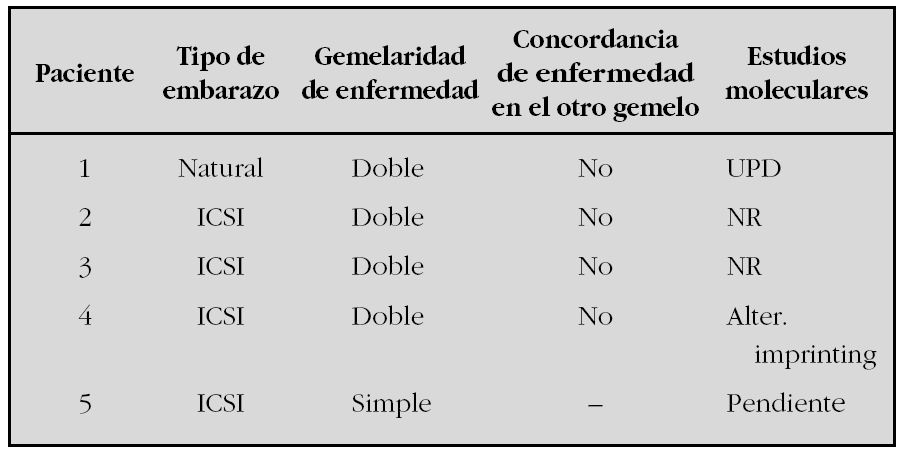
Los datos que observamos confirman las comunicaciones realizadas por otros registros e investigadores en poblaciones de pacientes con SBW en la que se ha observado un incremento de la gemelaridad y de pacientes nacidos con TRA.
Estos datos orientan a mecanismos epigenéticos intrínsecos probablemente asociados tanto a las técnicas de reproducción asistida como al proceso biológico de gemelaridad.
Síndrome otopalatodigital tipo 2 en 2 hermanos. Estudio autópsico y molecular
A. Mariño, P. Lapunzina, E. García-Fernández, E. González-Obeso y J.I. Rodríguez
Departamento de Anatomía Patológica y Servicio de Genética. Hospital Universitario La Paz. Madrid. España.
El síndrome otopalatodigital tipo 2 (OPD2) es una rara entidad con herencia recesiva ligada al cromosoma X caracterizada por una cara anormal hipoplasia centrofacial, hipertelorismo y paladar hendido estatura reducida, incurvación de huesos largos y anomalías óseas en pies y manos. Recientemente se ha demostrado que es debida a mutaciones en el gen de la filamina A (FLNA), también alterado en entidades alélicas como el síndrome OPD1, el síndrome Melnik-Needles y la displasia frontometafisaria. Se ha propuesto el término osteodisplasia frontootopalatodigital para agrupar estas cuatro entidades con características comunes.
Presentamos el caso de un recién nacido muerto de sexo masculino, macerado, de 33 semanas EG. La ecografía prenatal había detectado incurvación de fémur y húmero. En la autopsia encontramos anomalías características del OPD2: hipertelorismo, raíz nasal ancha, prominencia frontal, hipoplasia mediofacial, micrognatia, ausencia de paladar blando, tórax estrecho, espina bífida oculta dorsal, incurvaciones de huesos largos, ausencia de peronés y diversas anomalías esqueléticas en pies y manos. Se realizó consejo genético y seguimiento del segundo embarazo de la pareja con amniocentesis (46XY, normal) y ecografía fetal que en la semana 18 encontró arqueamiento de huesos largos con pies y manos anormales, por lo que se realizó IVE. Los tejidos fetales fueron estudiados anatomopatológicamente mostrando anomalías esqueléticas similares a las del primer hijo. Se extrajo ADN de los tejidos fetales y de la sangre de la madre, del padre, y de la abuela y tía maternas. El análisis de mutaciones demostró una transversión G629T en el gen FLNA en Xq28 del feto, mutación no descrita en la literatura especializada que estaba presente en heterozigosis en la madre. La abuela y la tía no mostraron alteraciones en el gen FLNA.
Este caso pone de manifiesto la importancia de la autopsia pediátrica, incluso en fetos macerados, para posibilitar un adecuado consejo genético.
Síndrome de Laurin-Sandrow: presentación de un nuevo caso
M. Álvarez, E. González-Obeso, V. Tarín, C. Morales, F. Omeñaca y J.I. Rodríguez
Departamento de Anatomía Patológica y Servicio de Neonatología. Hospital Universitario La Paz. Madrid. España.
El síndrome de Laurin-Sandrow se define como la tríada de polisindactilia en espejo de manos y pies y alteraciones nasales.
Presentamos el caso de un recién nacido de sexo femenino (46,XX), de 33 semanas de edad gestacional y 2.230 g, hijo de padres sanos no consanguíneos, con abombamiento frontal, hipertelorismo, nariz plana con hendidura en la columela y boca en "V" invertida con labios finos. Las manos tenían 7 dedos fusionados hasta las uñas y un apéndice postaxial carnoso redondeado. El pie derecho tenía 12 dedos y 11 el izquierdo, los cuatro externos eran reconocibles, el resto correspondía a una masa uniforme con uñas independientes. Había un apéndice digitiforme de 2,3 cm de longitud implantado en la parte interna de ambos pies. El estudio radiológico mostró siete metacarpianos y siete metatarsianos con morfología similar, las manos carecían de pulgar y en los pies los cuatro dedos externos tenían falanges regulares, mientras el resto tenía falanges irregulares. El apéndice digitiforme izquierdo tenía tres huesos y el derecho dos. Las tibias eran más cortas que los peronés.
En el sistema nervioso central (SNC) la oliva bulbar tenía morfología anómala, existían heterotopias cortical cerebelosa y de sustancia gris en cordón medular y en sustancia blanca hemisférica, marcada dilatación ventricular y gliosis de sustancia blanca.
Hay 10 casos de síndrome de Laurin-Sandrow descritos en la literatura médica. La novedad de este caso reside en que es el primero con estudio autópsico y en que presenta alteraciones en el SNC que no han sido descritas con anterioridad.
Síndrome Mietens-Weber: dos nuevos pacientes
V.M. Martínez-Glez, P. Lapunzina, A. Delicado, M.L. de Torres, M.A. Mori, M. Palomares, L. Fernández, J. Arcas e I. López Pajares
Servicio de Genética Médica. Hospital Universitario La Paz. Madrid. España.
Mietens y Weber (1966) describieron a 4 de 6 hermanos de padres consanguíneos, que mostraban retraso del crecimiento, dislocación de la cabeza del radio, contracción en flexión de ambos codos, cúbito y radio cortos, opacidad corneal bilateral, nistagmo horizontal y rotatorio, estrabismo, nariz pequeña y retraso mental de suave a moderado. Desde esta descripción, únicamente se han comunicado otros 4 pacientes. Presentamos dos nuevos casos en una pareja de gemelas de 6 años de edad.
Las pacientes, de padres jóvenes y no consanguíneos, nacieron después de un embarazo normal. Los hallazgos encontrados tras el nacimiento incluyen nistagmo horizontal y dislocación de los codos debido a un acortamiento anormal del cúbito y radio en ambas gemelas. Exploraciones clínicas posteriores revelaron un retraso psicomotor moderado con un marcado compromiso del lenguaje. Los cariotipos fueron normales.
Una revisión de la literatura especializada muestra que el síndrome de Mietens-Weber (OMIM: 249600) es un desorden infrecuente con un probable patrón de herencia autosómico recesivo. En lo que conocemos, incluyendo los 2 casos descritos aquí, solamente 10 casos han sido comunicados hasta ahora. Deseamos resaltar que el hallazgo de nistagmo congénito y la dislocación congénita de los radios en un paciente con retraso mental progresivo no es aleatorio y es altamente sugestivo del síndrome de Mietens-Weber.
Síndrome de retraso del crecimiento, dismorfias faciales y braquidactilia (síndrome de Frías). Presentación del segundo caso
M.ªL. Martínez-Frías 1,2, J. Fernández Toral 3, F. López-Grondona 1, E. Bermejo 1 y J. Mendioroz 1
1CEMC, Centro de Investigación sobre Anomalías Congénitas (CIAC). Instituto de Salud Carlos III. Madrid. 2Profesora del Departamento de Farmacología. Facultad de Medicina. Universidad Complutense de Madrid. 3Jefe de Sección de Genética Pediátrica. Hospital Universitario de Asturias. Profesor Titular de Pediatría de la Facultad de Medicina de la Universidad de Oviedo. España.
En el año 1975 Frías et al, describieron un niño de 6 años y medio que a partir del primer año de vida había mostrado una curva de crecimiento que se fue haciendo más lenta y con un desarrollo psicomotor también lento. En la escuela tenía un aprendizaje pobre y el estudio psicológico lo calificó como con "inteligencia por debajo de la media".
Cuando fue evaluado a los 6 años y medio, tenía una talla que correspondían a la cronológica de un niño de 4 años y 10 meses, y un peso adecuado a su talla. En la exploración clínica se detectaron: fisuras palpebrales antimongoloides con epicanto, hipertelorismo (distancia intercantal 3,1 cm) y ptosis palpebral. Los pabellones auriculares estaban rotados y con forma de copa. Las manos mostraban dedos cortos, desviación cubital de los dedos índices y clinodactilia de los 5.º dedos. En los pies, los dedos también eran cortos. La exploración con rayos X mostró que las falanges medias de los dedos segundo, tercero, cuarto y quinto eran muy pequeñas, aunque el dedo tercero era el menos afectado. Defectos similares se apreciaron en los dedos segundo a quinto de los pies. La edad ósea estaba retrasada correspondiendo a la cronológica de un niño con 4 años y medio. La madre del niño, tenía una estatura baja y sobrepeso, con un patrón de anomalías idéntico al de su hijo. Los autores consideraron que este patrón de alteraciones constituía un nuevo síndrome autosómico dominante.
Presentamos una familia de tres generaciones en la que se han identificado 4 personas con un fenotipo concordante con el descrito por Frías et al (1975), aunque con cierto grado de variabilidad. La propositus tiene un cariotipo de alta resolución (850 bandas) normal. Tras la revisión de la literatura médica, esta es la segunda familia descrita con este patrón de defectos, que confirma tanto el síndrome de Frías como su modelo de herencia autosómico dominante.
Este trabajo se ha realizado dentro del Programa "Redes Temáticas de Investigación Cooperativa" del Instituto de Salud Carlos III. Ministerio de Sanidad y Consumo. Expte.: G03/123. REpIER. Y, en parte, por la "Fundación 1000" sobre defectos congénitos.
La hemizigosidad para NCF1 parece proteger frente al desarrollo de HTA en pacientes con síndrome de Williams
M. Del Campo, L.F. Magano, A. Antonell, F. Muñoz, R. Flores, M. Bayes y L.A. Pérez Jurado
Unidad de Genética. Unidad de Transducción de Señales. Departamento de Ciencias Experimentales y de la Salud. Universitat Pompeu Fabra. Barcelona. España.
El síndrome de Williams-Beuren (SWB) es un trastorno del desarrollo con fenotipo multisistémico causado por una aneusomía parcial dentro de la banda cromosómica 7q11.23. Se sabe que la deleción del gen de la elastina es la causa de las estenosis vasculares y se piensa que esto es lo que predispone al desarrollo de hipertensión arterial (HTA) en aproximadamente 50 % de pacientes. La deleción más común de ~1,55 Mb está mediada por recombinación homóloga no alélica entre grandes bloques de duplicaciones segmentarias. Estos bloques contienen genes funcionales y seudogenes, específicamente varias copias de dos genes (NCF1 y GTF2IRD2), cuyo número de copias activas varía en función de la localización del punto de rotura de la deleción. Realizamos el mapeado preciso de las deleciones por medio del análisis de variantes de copias parálogas para investigar la asociación de potencial de los diversos rasgos fenotípicos con la extensión de la deleción. La muestra incluyó 100 pacientes esporádicos con deleciones de similar tamaño en 7q11.23. Se encontraron asociaciones significativas entre los diversos rasgos clínicos y los diferentes subtipos de deleción. En particular, se determinó que la HTA, no asociada a la presencia o gravedad de lesiones estenóticas cardiovasculares objetivables, sí era más frecuente en pacientes sin deleción del gen NCF1 (p < 0,005). NCF1 codifica para una subunidad citosólica de la nicotinamida adenindinucleótido fosfato reducido (NADPH) oxidasa leucocitaria (p47PHOX), un complejo enzimático que cataliza la producción de superóxido (O2) a partir de NADPH y oxígeno. Con el objetivo de conocer el mecanismo molecular por el cual la dosis génica de NCF1se correlaciona con la tensión arterial, determinamos por Western Blot que sí existía una reducción significativa de la cantidad de proteína p47PHOX en pacientes hemizigotos para este gen, y comparamos la activación de la cascada respiratoria (test de tetrazoilo de azul nitroso) tras el tratamiento con acetato miristato de forbol (1?M) en líneas celulares de pacientes con SWB con y sin deleción del gen NCF1. Como esperábamos, las pacientes hemizigotas para el gen NCF1 mostraron una menor actividad de la NADPH oxidasa y consecuentemente una menor producción de superóxido (p < 0,005).
Estos hallazgos sugieren que la deleción de NCF1 determina una reducción de la capacidad de generar estrés oxidativo y así protege contra el desarrollo de HTA a la cual están predispuestos los pacientes con síndrome de Williams. Este hecho sugiere que la terapia antioxidante puede estar indicada en este grupo de pacientes para tratar y/o prevenir la HTA.
Mosaicismo para una deleción de 8 mb en 7q11.2 en un paciente con síndrome de Williams y rasgos autistas
L.A. Pérez-Jurado 1, S. Castillo 2, L.F. Magano 1, R. Flores 1, X. Carrasco 3, P. Rothhammer 4, M. Aracena 5, V. Daher 3, M. del Campo 1 y F. Aboitiz 4
1Unitat de Genètica. Universitat Pompeu Fabra. Barcelona. España. 2Sección de Genética. Hospital Clínico. 3Instituto de Ciencias Biomédicas. Facultad de Medicina. 4Departamento de Psiquiatría. Pontificia Universidad Católica de Chile. 5Servicio de Genética. Hospital Luis Calvo Mackenna. Universidad de Chile. Santiago de Chile.
Presentamos un varón chileno de 5 años de edad con rasgos clínicos de síndrome de Williams (aspecto facial, estenosis aórtica supravalvular, hernias inguinales, voz ronca) asociados a un retraso psicomotor severo con ausencia de lenguaje, características autistas y heterocromía parcial del iris izquierdo. Mediante estudios repetidos de hibridación in situ con fluorescencia (FISH) en muestras de sangre periférica, se detectó una deleción de la sonda de ELN con un patrón en mosaico (deleción en 41-46 % de metafases). El estudio de muestras de ADN de diversos tejidos (sangre, mucosa bucal, bulbo piloso), demostró herencia biparental con una reducción relativa de dosis del alelo paterno muy variable entre tejidos, oscilando desde el 60 % en células hemáticas de origen mesodérmico hasta el < 1 % en células de bulbo piloso, de origen ectodérmico. El cartografiado de la deleción mediante microsatélites, sondas de copia única y variantes parálogas de secuencia en la muestra de pelo, reveló una deleción de ~8 Mb en 7q11.2 con puntos de rotura localizados en duplicaciones segmentarias específicas del cromosoma 7 que se extienden 26,8 kb con un 98,5 % de identidad de secuencia. Además de la región crítica completa del síndrome de Williams, la deleción incluye al menos cuatro genes conocidos de copia única: AUTS2, WBSCR17, CALN1 y WBSCR16. El fenotipo neuroconductual severo con rasgos autistas de este paciente puede ser debido, al menos en parte, a haploinsuficiencia para AUTS2, un gen que se ha encontrado previamente alterado por una translocación en 2 hermanos con autismo primario. Este estudio ilustra que la recombinación homóloga no alélica mediada por duplicaciones segmentarias también ocurre en mitosis, y que hay que considerar la posibilidad de mosaicismo para una deleción en pacientes con claro fenotipo y estudio citogenético-molecular aparentemente negativo.
Identificación de deleciones de amplio rango en el extremo distal 39 del genSHOXen pacientes con discondrosteosis de Léri-Weill (DLW)
S. Benito Sanz, D. Gorbenko del Blanco, M. Aza Carmona, A. Campos Barros A, J. Argenté y K.E. Heath
Hospital Niño Jesús. Madrid. España.
El gen SHOX, localizado en la región seudoautosómica (PAR1) de los cromosomas X e Y, codifica una proteína homeodominio que actúa como factor de transcripción en la regulación del crecimiento esquelético. Las mutaciones del gen SHOX o deleciones parciales de los cromosomas X e Y que afecten al gen SHOX, producen haploinsuficiencia de SHOX asociada con un fenotipo variable de talla baja junto con anomalías esqueléticas características de la discondrosteosis de Léri-Weill (DLW), displasia mesomélica de Langer (DML) y talla baja idiopática (TBI). Se ha estimado que ~50 % de los pacientes con DLW exhiben una anomalía en el gen SHOX, mientras que en el 50 % restante no se han detectado alteraciones en dicho gen, desconociéndose el defecto molecular causante de la talla baja y la discondrosteosis.
Estudios recientes en nuestro laboratorio han podido detectar, sin embargo, la presencia de deleciones de amplio rango en el extremo distal 39 del gen SHOX en una familia multigeneracional con DLW. En todos los miembros afectados la deleción, aún sin afectar al gen SHOX, segrega con el fenotipo, lo que sugiere la existencia bien de un posible locus distal de control transcripcional de SHOX o bien de un locus alternativo en la región PAR1 que, independientemente de SHOX, tiene un efecto modulador o regulador del crecimiento.
Con ayuda del programa "Tandem Repeat Finder" (http://c3. biomath.mssm.edu/trf.html), hemos diseñado un nuevo panel de microsatélites marcadores en la región PAR1 que nos ha permitido cartografiar y delimitar la amplitud de la secuencia genómica de la región PAR1 afectada por la deleción con el fin de identificar la región donde se encuentra el posible locus de control transcripcional del gen SHOX o locus alternativo involucrado (karen.heath@uam.es).
Síndromes cardiomiélicos: caracterización de una nueva mutación en un paciente con síndrome de Holt-Oram
L. Fernández 1, P. Lapunzina 1, A. Delicado 1, A. Sharif 2, G.S. Cross 2, M.A. Mori 1, M.L. de Torres 1, M. Palomares 1 y I. López Pajares 1
1Servicio de Genética Médica. Hospital Universitario La Paz. Madrid. España. 2Department of Molecular Genetics. Centre for Medical Genetics. Nottingham City Hospital. United Kingdom.
Los síndromes cardiomiélicos comprenden cardiopatías congénitas y malformaciones esqueléticas de los miembros superiores, y están relacionados con mutaciones deletéreas de factores de transcripción con dominios del tipo T-Box. El síndrome de Holt-Oram se debe a una mutación dominante en el gen TBX5 que altera la estructura tridimensional de la proteína impidiendo su correcta unión al ADN. Se han descrito varias mutaciones puntuales y deleciones del gen TBX5 en pacientes con fenotipo de Holt-Oram.
El propósito es un niño con una CIA ostium secundum grande y una CIV diagnosticados por clínica (soplo) y ecocardiografía. Presenta además un emplazamiento distal bilateral de los dedos pulgares, con un índice de implantación de 0,19 frente a una media normal de 0,50 para su edad gestacional al nacer. Es remitido a la consulta de genética para descartar microdeleción 22q11.2. El cariotipo y la hibridación in situ con fluorescencia (FISH) con sonda D22S75 resultaron normales y debido a los hallazgos clínicos se envió para estudio molecular del síndrome de Holt-Oram, encontrándose una mutación en el exón 7 de TBX5 que produce una probable alteración del splicing del gen que da lugar a una proteína truncada en su extremo C terminal. Los padres del propósito presentan una secuencia normal para el gen, lo que indica que la mutación se produjo de novo, sin que pueda descartarse un mosaicismo germinal en los padres.
El síndrome de Holt-Oram es una enfermedad relativamente común y la causa más frecuente de síndrome cardiomiélico. Debe ser objeto de estudio molecular todo niño con malformaciones cardíacas y alteraciones de las extremidades superiores como pulgares ausentes, distalmente emplazados o trifalángicos.
Anomalías craneofaciales atípicas en la trisomía 13
E. García-Fernández, C. Morales, M.A. Mori, A. Mariño, E. González-Obeso y J.I. Rodríguez
Departamento de Anatomía Patológica y Servicio de Genética. Hospital Universitario La Paz. Madrid. España.
La trisomía 13 es un síndrome que combina anomalías craneofaciales características con malformaciones cardíacas, urogenitales, esqueléticas y del sistema nervioso central (SNC). Existe un grado variable de holoprosencefalia en más de dos tercios de los casos, que condiciona anomalías faciales con dos espectros fenotípicos diferentes: unos tienen desde hipotelorismo a ciclopía y otros hendiduras labiopalatinas con labio leporino lateral, central o agenesia de premaxila.
Presentamos 3 casos de trisomía 13 con un fenotipo facial diferente de los "clásicos" en este síndrome. La cara tosca y redondeada, la pirámide nasal muy grande, el occipucio plano, una marcada micrognatia y unas orejas con plegamiento anormal y hélix fino caracterizaban a los 3 recién nacidos. Ninguno de ellos presentaba frente huida, hipotelorismo ni labio leporino.
Sin embargo, no diferían en cuanto a las malformaciones internas. En las autopsias se encontró cardiopatía congénita (dos con tetralogía de Fallot y uno con CIA y CIV), anomalías genitales (útero bicorne en las 2 niñas y criptorquidia en el niño), renales (riñones trisómicos en todos), esqueléticas (polidactilia postaxial en dos y 10 costillas en las tres) y, en el SNC, agenesia de los bulbos olfatorios y heteropias corticales cerebelosas en todos, y microftalmía en dos de ellos.
Al revisar la literatura médica no hemos encontrado descrito este fenotipo atípico en la trisomía 13.
Síndrome de Fraser
A.M.ª Cueto 1, T. Vendrell 1, A. Bonato 2 y N. Toran 3
Unidades de 1Genética, 2Medicina Fetal y 3Anatomía Patológica. Hospital Materno-Infantil Vall d'Hebron. Barcelona. España.
El síndrome de Fraser se caracteriza por criptoftalmos, sindactilia cutánea, malformaciones laríngeas y del tracto genitourinario, dismorfias craneofaciales y anomalías musculoesqueléticas. Su herencia es autosómica recesiva, genéticamente heterogéneo y sólo se han detectado mutaciones en el gen FS1 en población específica.
El primer caso es una quinta gestación (21.4 SG). Por ecografía se objetiva agenesia renal bilateral, anhidramnios, extremidades muy flexionadas y edema nucal. A la exploración destaca criptoftalmos izquierdo, raíz nasal amplia con punta bífida, fisura de paladar posterior, orejas displásicas, braquisindactilia cutánea de manos y pies, genitales ambiguos, agenesia renal bilateral, hipertrofia de miocardio izquierdo, hipoplasia pulmonar bilateral, vejiga urinaria hipoplásica y flexión de las 4 extremidades. Cariotipo: 46,XY. Se orienta como probable síndrome de Fraser. Pendiente de estudio molecular.
El segundo caso es una tercera gestación (19 SG) de padres consanguíneos con su pareja. Refiere 2 embarazos anteriores: una interrupción del embarazo a las 20 SG con oligoamnios, agenesia renal bilateral, sindactilia de manos y pulmones de gran tamaño. No se orientó su etiología. El segundo embarazo fue un aborto espontáneo (12 SG). En la gestación actual se objetiva en la ecografía, oligoamnios, pulmones hiperrefringentes bilaterales y anomalías renales y de extremidades. A la exploración destaca criptoftalmos derecho, raíz nasal amplia, orejas displásicas, cuello corto, tórax amplio, abdomen globuloso, sindactilia cutánea parcial de manos y pies, genitales ambiguos e hipoplásicos, hipoplasia de epiglotis y estenosis laríngea extrema, hiperexpansión pulmonar bilateral, malrotación intestinal, agenesia renal bilateral, vejiga urinaria hipoplásica, atresia anal, útero hipoplásico y ovarios de aspecto disgenético. Cariotipo: 46,XX. Se orienta como probable síndrome de Fraser y se relaciona con el primer embarazo y con otros familiares afectados. Se realiza estudio molecular.
Aproximación al diagnóstico clínico de las craneosinostosis
M.D. Saavedra 1, M. Orera 2, J. Romero 3 y J. Fernández 4
1Fundación Investigación. 2Unidad de Genética. 3Servicio de Radiología. Hospital General Universitario Gregorio Marañón. 4Universidad Europea de Madrid. España.
Introducción: Las craneosinostosis se producen por el cierre prematuro de una o más líneas de sutura entre los huesos que forman el cráneo. Las suturas se cierran precozmente por sinostosis activa primaria produciéndose cambios no sólo en las estructuras óseas vecinas a la sutura osificada, sino también en áreas distantes, ya que el crecimiento óseo se detiene perpendicularmente a la sutura osificada, existiendo crecimiento compensatorio o sobrecrecimiento en sentido paralelo a la estenosis. Así, la morfología craneal (plagiocefalia, braquicefalia, turricefalia, acrocefalia, cráneo en trébol, etc.) está determinada por el orden y velocidad de progresión del cierre de las suturas osificadas, ya que los tejidos blandos, primordialmente el cerebro, tienden a crecer por las áreas libres que dejan las suturas cerradas, causando dismorfias secundarias y desproporciones ente las diversas estructuras que forman en macizo craneofacial. En conjunto las craneosinostosis tienen una frecuencia de 1/2.500 y se presentan con una gran variabilidad clínica en ocasiones asociada a graves complicaciones.
Más del 90 % de las craneosinostosis son de origen genético y aparecen en general con un patrón de herencia autosómico dominante.
Objetivo: Estandarización del diagnóstico clínico de las craneosinostosis para: confirmar los diagnósticos de sospecha, identificar la entidad nosológica presente, determinar la presencia/ausencia de la enfermedad en los familiares de los afectados y poder calcular los riesgos de recurrencia en cada familia.
Material y métodos: Pacientes del Hospital General Universitario Gregorio Marañón, con dismorfias craneofaciales, que son enviados para su evaluación a la Unidad de Genética Pediátrica. Se realiza el árbol genealógico y la exploración clínica del paciente y de sus padres y hermanos. Posteriormente se realiza una cefalometría por la misma persona en idénticas condiciones, estandarizadas para este fin. Las cefalometrías se digitalizan y son trazadas, medidas e interpretadas por la misma persona. Los datos son analizados estadísticamente, usando los métodos de Riketts, Steinert y McNamara y se comparan con los estándares de normalidad para la población española.
Resultados: El método propuesto se ha utilizado en 10 pacientes pediátricos con sospecha de craneosinostosis. En todos ellos se confirmó la presencia de la misma y la existencia de un progenitor afectado.
Agradecimientos: Trabajo financiado por la Red Temática de Investigación Cooperativa, Genética Clínica y Molecular y la Universidad Europea de Madrid.
Síndrome MICRO: descripción de un nuevo paciente con seguimiento prolongado
I. Arroyo Carrera 1, M.L. Martínez-Frías 2, A. 1 López Lafuente, M.J.García García 1 y L. Rodríguez Martínez 2
1Unidad de Neonatología. Hospital San Pedro de Alcántara. Servicio Extremeño de Salud. Cáceres. 2ECEMC. Centro de Investigación sobre Anomalías Congénitas. Instituto de Salud Carlos III. Madrid. España.
El síndrome MICRO es un patrón autosómico recesivo de anomalías congénitas caracterizado por microcefalia, displasia cortical, microcórnea, cataratas, atrofia óptica, hipoplasia del cuerpo calloso, retraso mental profundo, diplejía hipotónica e hipogenitalismo. Desde el año 1993 en que se publicó la primera familia donde se identificó el síndrome han aparecido al menos siete publicaciones más con descripción de nuevos pacientes, una de ellas con 2 casos españoles.
Presentamos un paciente con manifestaciones clínicas compatibles con el síndrome al que hemos realizado seguimiento desde el nacimiento hasta la actualidad, 13 años y medio.
Caso clínico: Varón, producto de primera gestación, madre 28 años, padre 26, no consanguíneos, no historia familiar. Edad gestacional 38 semanas. Apgar 19 = 8, 59 = 9. Peso al nacimiento 2.100 g, talla 43 cm, perímetro cefálico 30 cm, todos inferiores a P10. Fenotipo facial no característico con narinas antevertidas, nevo en dorso de la nariz, microcórneas, catarata congénita derecha, fisura palatina en los dos tercios posteriores de la línea media, microrretrognatia y desarrollo posterior de asimetría facial con hemihipoplasia izquierda; cuello corto, pliegue palmar único transverso bilateral, sindactilia cutánea de segundo y tercer dedos de los pies que afecta a primera y segunda falanges del pie derecho y primera del pie izquierdo, arteria umbilical única, criptorquidia bilateral, hernia inguinal derecha, comunicación interventricular subaórtica con repercusión hemodinámica que precisó cirugía a los 2 años, agenesia renal izquierda. En su evolución microcefalia con perímetro cefálico 48,5 cm a los 13 años, neurológicamente retraso mental profundo sin marcha ni sedestación estable ni lenguaje excepto emisión de gritos, epilepsia (primera crisis a los 12 meses) con mal control pese a politerapia, se realizan tratamientos rehabilitadores y ortopédicos desde edad muy temprana para prevención contracturas. RM cerebral: agenesia del cuerpo calloso. Atrofia óptica bilateral con PEV: ojo derecho 162,9 ms, potencial muy retrasado casi abolido, ojo izquierdo 141,8 ms, potencial muy retrasado. Se realizó al nacimiento serología infección connatal negativa y posteriormente, cuando se describió, estudio del metabolismo del colesterol para descartar síndrome de Smith-Lemli-Opitz con resultado normal. Cariotipo de alta resolución (600 bandas) 46,XY.
Discusión: Al valorar pacientes con el grupo de manifestaciones clínicas que incluyen microcefalia congénita, microcórnea y cataratas debemos pensar en el síndrome cerebro-óculo-facio-esquelético y en el síndrome de Cockayne, en ambos síndromes existe una hipersensibilidad a la radiación ultravioleta objetivable en cultivo celular de fibroblastos no existente en el síndrome MICRO, esta prueba está pendiente de realizar en nuestro caso. También, además del síndrome de Smith-Lemli-Opitz, debemos considerar el síndrome de Martsolf y el síndrome de cataratas congénitas-dismorfia facial-neuropatía.
Creemos que las manifestaciones clínicas oftalmológicas, neurológicas y genitales de nuestro paciente son compatibles con las descritas en el síndrome MICRO. Queremos resaltar las otras manifestaciones clínicas también presentes en nuestro paciente: cardíacas, renales, fisura palatina, arteria umbilical única, no descritas o sólo de forma aislada en la literatura médica, porque pensamos deben incorporarse al espectro fenotípico del síndrome y tal vez hayan sido infradiagnosticadas en los casos publicados.
Fracturas congénitas: una presentación clínica inusual de la enfermedad de Menkes
C. Vázquez 1, J.J. Alcón 1, L. Birk Moller 2, N. Horn 2, C. Díaz 1, H. Cortina 1 y A. Pérez Aytes 1
1Hospital Infantil La Fe. Valencia. España. 2Instituto J.F. Kennedy. Glostrup. Dinamarca.
La enfermedad de Menkes (MIM: 309400) es un trastorno en el metabolismo del cobre ligado a X recesivo (Xq13.3, gen: ATP7A). La presentación habitual es el deterioro neurológico progresivo en los primeros meses de vida. Presentamos un caso de enfermedad de Menkes que se inició con fractura craneal congénita.
Caso clínico: R.P. es un varón, tercera gestación en una madre de origen español, 25 años, sana. Primera gestación un aborto espontáneo, la segunda una niña que vive sana. Parto en la 38 semana, cefálico, acabado por cesárea. Extracción fetal limpia, no traumática. Apgar 9/10. Se aprecia crepitación en zona temporal y parietooccipital. Es remitido de urgencia a nuestro Hospital por fractura craneal congénita con hematoma epidural. Fue intervenido por Neurocirugía y una vez recuperado se observó hipotonía así como fracturas múltiples (húmero derecho, peroné y tibia izquierda), piel muy redundante y cabello fino y escaso. Se pensó en osteogénesis imperfecta pero los signos radiológicos de cráneo y huesos largos no eran compatibles con este diagnóstico. Dentro de los estudios bioquímicos se pidió cobre en sangre pensando en la enfermedad de Menkes. Las cifras de cobre estaban por debajo del rango normal iniciándose tratamiento con histidinato de cobre subcutáneo. En fibroblastos de piel (laboratorio Dra. N. Horn) se observó aumento significativo en la captación de 64Cu lo que confirmó la enfermedad de Menkes. El estudio de genética molecular identificó una mutación en el lugar de ensamblaje del exón 14 (IVS14 + 1G > A) del gen ATP7A.
Comentario: Aunque en el proceso evolutivo de la enfermedad de Menkes pueden aparecer fracturas óseas como parte del cuadro clínico, la presentación de fracturas congénitas es extremadamente rara. Hemos encontrado únicamente dos referencias de casos similares en la literatura especializada. La grave deficiencia en el metabolismo del cobre produce anomalías en diversas enzimas dependientes, entre ellas la lisil-oxidasa, una enzima necesaria para el metabolismo del colágeno, lo que explicaría los trastornos óseos que se producen en estos pacientes. En nuestro caso la mutación IVS14 + 1G > A no estaba presente en la madre ni en la abuela materna por lo que se asume una mutación de novo.
Aranodactilia contractural congénita. Una nueva familia
E. Galán Gómez 1,2, M. García Reimundo 2, P. García Tamayo 2, J.M.ª Carbonell Pérez 1, J.J. Cardesa García 2 y M.ªL. Martínez Frías 3
1Unidad de Genética. Unidad de Prevención de Minusvalías. 2Departamento de Pediatría. Hospital Materno-Infantil de Badajoz. Universidad de Extremadura. Servicio Extremeño de Salud. Junta de Extremadura. Badajoz. 3CIAC. Instituto de Salud Carlos III. Madrid. España.
Presentamos un recién nacido varón de 2 días de edad que consulta por presentar dedos largos, contracturas articulares y anomalías de pabellones auriculares.
Antecedentes: a) Familiares: madre con escasa masa muscular, dedos largos y contracturas articulares. Un hermano de la madre pretérmino, falleció a las 2 h de vida, con fenotipo similar al paciente. Abuelo materno, 1 hermano y 3 hermanas del mismo, y el padre y abuela materna de este último afectados de contracturas y aranodactilia con diferente gravedad clínica. b) Personales: producto de primer embarazo, hijo de padres de edades de 31 y 34 años (edad materna y paterna, respectivamente) en la gestación. La madre padeció proceso gripal sin fiebre a las 16 semanas de edad gestacional y fue tratada con amoxicilina. Parto a las 39 semanas por fórceps. Sin reanimación al nacer. Peso al nacer de 3.150 g.
Exploración: normocefalia, hendiduras palpebrales horizontales, ojos normales, ligera micrognatia, pabellones auriculares de implantación normal, grandes, blandos y laxos con lóbulos con pliegues y arrugados. Contractura a nivel de musculatura izquierda del cuello. Tórax normal. Soplo protones. 1/6. Abdomen normal. Extremidades: a) Superiores: contracturas en flexión de codos, contracturas de dedos de primera y segunda articulaciones interfalángicas. Dedos largos, escasos pliegues en dedos. b) Inferiores: contracturas en flexión en rodillas. Ambos pies en valgos. Dedos de pies largos. Genitales: normales de varón.
Exámenes complementarios: estudio cardiológico normal. Cariotipo en sangre periférica por técnica convencional y bandas GTG (resolución 600 bandas) normal. Cariotipo 46,XY.
Se presenta el caso y se revisa la literatura médica.
Hiperfosfatasia infantil: diagnóstico diferencial en la fragilidad ósea del niño
A. González-Meneses López, F. Yanes Sosa, G. Rodríguez Criado y J. Martín Govantes
Unidad de Dismorfología. Hospitales Universitarios Virgen del Rocío. Sevilla. España.
Introducción: La hiperfosfatasia es una enfermedad que afecta a la incorporación del calcio a la matriz ósea. Raramente se manifiesta en la infancia, donde provoca un anormal modelado óseo, y un aumento de su fragilidad, condicionando deformidades y fracturas múltiples.
Caso clínico: Varón de 2 años de edad remitido por sospecha de osteogénesis imperfecta, que presentaba diversas fracturas de miembros con cifosis dorsal y osteopenia intensa, junto a un anormal modelado óseo, especialmente de los huesos largos.
Analíticamente destacaba un nivel de fosfatasa alcalina de 4.000 U/dl con normalidad en el resto de parámetros del metabolismo fosfocálcico y funcionamiento renal. Se descartó la presencia de una enfermedad de depósito, y la biopsia ósea mostró signos de elevado recambio óseo compatible con el diagnóstico de sospecha de hiperfosfatasia. No presenta antecedentes familiares de osteopenia o deformidad ósea. La confirmación del proceso por biología molecular se encuentra en curso. Se ha comenzado tratamiento con difosfonatos parenterales con buena tolerancia.
Comentarios: Ante un cuadro de fragilidad ósea en la infancia se debe realizar un diagnóstico diferencial con los procesos que más frecuentemente se manifiestan de esta manera, especialmente con la osteogénesis imperfecta en sus distintas formas y con las alteraciones del metabolismo fosfocálcico. En ocasiones, ciertas displasias óseas pueden simular un cuadro similar.
La importancia de un diagnóstico lo más precoz posible viene determinada por el mejor resultado del tratamiento con difosfonatos en la calidad de vida del paciente.
Hipofosfatemia ligada al cromosoma X: descripción de nueva mutación en el gen PHEX
F.J. Ramos 1, I. Bueno 1, G. Bueno 2, T. Strom 3 y M. Bueno 1
1Sección de Genética y Endocrinología. 2Departamento de Pediatría. Hospital Clínico Universitario. Facultad de Medicina. Universidad de Zaragoza. España. 3Institut Humangenetik. Technischen Universität. München. Alemania.
La hipofosfatemia ligada al cromosoma X (HLX) (OMIM: 307800), también denominado raquitismo resistente a la vitamina D, es una enfermedad hereditaria causada por un defecto en la reabsorción tubular renal de fosfatos cuyo gen (PHEX) se localiza en el brazo corto del cromosoma X (Xp22). Clínicamente se caracteriza por raquitismo insensible a la vitamina D, retraso del crecimiento, anomalías óseas y defectos de la dentición. Los individuos afectados presentan hipofosfatemia, debida a la deficiente reabsorción renal de fosfatos y niveles bajos de vitamina D (1,25-OH) en suero. Los niveles séricos de calcio, PTH y 25-OH son normales. En general, ambos sexos están igualmente afectados en la infancia, algo inusual en este tipo de herencia.
Presentamos una familia con HLX en la que hay 5 individuos afectados en 3 generaciones. La probando, de 2 años, consultó por posible displasia ósea, presentando baja talla (P3) y deformidades en extremidades inferiores. Sin embargo los datos bioquímicos eran compatibles con raquitismo hipofosfatémico: fosfatemia 2,6 mg/dl, calcemia 9,9 mg/dl, aunque el 1,25-OH sérico era normal (65 pg/ml) El estudio radiológico del esqueleto demostraba anomalías por deformidad en epífisis y metáfisis de huesos largos, con incurvación de diáfisis en extremidades inferiores. La historia familiar mostraba las mismas deformidades en la madre, 2 tías maternas y abuela materna, quienes habían sufrido diversas intervenciones quirúrgicas correctoras.
Considerando la posibilidad de HLX, se estudió el gen PHEX amplificando sus 22 exones y secuenciando en su totalidad las regiones codificantes, encontrándose en la paciente una mutación en heterozigosis por cambio de sentido (1936G > C, D646H) en el exón 19. Esta mutación no ha sido descrita previamente en la literatura médica y estaba presente en todos los demás miembros afectados, confirmándose así el diagnóstico de HLX. Se discutirá la correlación fenotipo-genotipo en el HLX.
Todos los autores españoles son investigadores de la Red Temática "RECGEN" (Ref. C03/07); I.B. y F.J.R. son investigadores de la Red "GIRMOGEN" (Ref. G03/098), ambas financiadas por el Ministerio de Sanidad y Consumo (FIS).
Secuencia de bridas amnióticas
A. Sanchis, A. de la Mano, A. Clement, J.L. Tortajada y C. del Castillo
Servicio de Pediatría. Hospital Universitario Dr. Peset. Valencia. España.
La rotura del amnios se relaciona con la formación de bridas amnióticas que rodearían estructuras fetales en desarrollo (generalmente miembros), provocando constricciones anulares, deformidades, amputación de partes distales, habitualmente asimétricas y constricción del cordón umbilical. Además, pueden producirse deformaciones secundarias a la disminución de la motilidad fetal, completando el cuadro conocido como secuencia de bridas amnióticas o secuencia ADAM (deformidad amniótica, adhesión, mutilación).
La mitad de los casos afectan a la parte distal de los miembros, pudiendo asociar acrania, cefalocele, coloboma, hendiduras faciales atípicas y defectos de la pared corporal, aunque estudios recientes indican que los casos con estos últimos sería una entidad diferente de origen más precoz.
Su origen es desconocido, esporádico y de baja recurrencia, relacionado con traumatismos y procedimientos fetales invasivos (biopsia de vellosidades coriales, amniocentesis), consumo de drogas (metadona, misoprostol), fiebre en primer trimestre y poblaciones viviendo a grandes altitudes.
Su prevalencia oscila entre 1/1.200-1/15.000 recién nacidos vivos (RNV), aumentando en los mortinatos. EL ECEMC obtiene una prevalencia de 1/21.163 RNV.
El mecanismo patogénico más aceptado es la rotura del amnios, generalmente antes de las 12 semanas, cuando el corion y el amnios son membranas separadas. Se han desarrollado otras teorías como el mecanismo disruptivo vascular o la alteración en la programación embrionaria en las fases tempranas de la morfogénesis.
El diagnóstico se realiza por la sintomatología y el estudio placentario, siendo posible el diagnóstico ecográfico prenatal, así como procedimientos quirúrgicos prenatales para mantener el flujo sanguíneo distal.
Presentamos un recién nacido sin antecedentes familiares de malformación ni hábitos tóxicos maternos. Al nacimiento se detectan defectos transversos distales de miembros, con reducción, amputación y deformidad de dedos. En la inspección placentaria se observa banda amniótica en región cercana a la inserción del cordón. Antecedente de amniocentesis en la semana 16 de gestación con cariotipo XY.
Síndrome de microcefalia-linfedema: herencia autosómica dominante frente a dominante ligada al cromosoma X
I. Bueno 1, M.P. Samper 2, P. Ventura 2, J. Pérez-González 2 y F.J. Ramos 1
1Sección de Genética y Servicio de Neonatología. 2Departamento de Pediatría. Hospital Clínico Universitario. Facultad de Medicina. Universidad de Zaragoza. España.
El síndrome de microcefalia-linfedema (SML) es una entidad autosómica dominante cuya primera descripción se debe a Leung et al, quienes, en 1985, describieron 5 individuos en cuatro generaciones de una misma familia china. La asociación de microcefalia aislada o de linfedema y displasia coriorretiniana descrita posteriormente en algunos pacientes, planteó cierta controversia sobre si se trataba de un único síndrome con expresión variable o de entidades diferentes. La descripción de familias con un patrón de herencia autosómico recesivo y ligado al cromosoma X incrementó la discusión.
Presentamos 2 hermanos varones con SML cuya madre tiene microcefalia. El menor presentaba al nacimiento un perímetro cefálico normal, pero desarrolló una microcefalia posnatal (DE < 2). Ambos hermanos presentan anomalías cardíacas, el mayor comunicación interauricular (CIA) e interventricular (CIV), y el pequeño insuficiencia tricuspídea. Ninguno presentaba coriorretinopatía, aunque el mayor desarrolló una hipermetropía severa a partir del año de vida. Ambos hermanos presentan actualmente un retraso psicomotor y pondoestatural. La madre tiene un astigmatismo grave, ECG normal y talla en el percentil 25.
En relación al mecanismo hereditario del SML en esta familia se plantea la posibilidad de herencia dominante ligada al cromosoma X frente a autosómica dominante con expresividad variable.
Todos los autores son investigadores de la Red Temática "RECGEN" (Ref. C03/07); I.B. y F.J.R. son investigadores de la Red "GIRMOGEN" (Ref. G03/098), ambas financiadas por el Ministerio de Sanidad y Consumo (FIS).
Síndrome de Feingold. A propósito de 2 familias
E. Gabau 1, C. Escofet 2, J. Artigas 2 e I. Lorente 2
Servicio de Medicina Pediátrica. Unidades de 1Genética Clínica y 2Neurología Pediátrica. Hospital de Sabadell. Corporación Sanitaria Parc Taulí. Sabadell. España.
El síndrome de Feingold se caracteriza por la presencia de microcefalia asociada a anomalías de extremidades que incluyen clinodactilia del segundo y quinto dedos de las manos y sindactilia del tercero, así como del cuarto y quinto dedo de los pies. Una tercera parte de los pacientes presentan atresia de esófago o del duodeno o de ambos. Dificultades para el aprendizaje o incluso retraso mental se presentan en el 80 % de los individuos. Es una condición de transmisión autosómica dominante.
Presentamos 2 familias no relacionas entre si. Familia 1: Julia de 3 años de edad es visitada en nuestra Unidad de Genética al presentar microcefalia y retraso de lenguaje. El antecedente de intervención quirúrgica neonatal por atresia de esófago junto con dismorfia facial, clinodactilia de segundo y quinto dedos de ambas manos y sindactilia 2-3 dedos de los pies. Nos permitió reconocer el síndrome de Feingold, que también presenta el padre y otros miembros de la familia paterna, aquellos miembros con microcefalia y clinodactilia presentan un cierto grado de retraso mental, según refiere el padre de Julia. No hay ningún otro miembro afectado de atresia esófago/duodeno. Familia 2: Daniel y Alba de 14 y 8 años, son hermanos y los visitamos en la consulta de Genética remitidos desde Neurología por microcefalia familiar y retraso mental, que también presenta la madre, tienen una hermana de 10 años sin problemas. En la exploración destaca clinodactilia de segundo y cuarto dedos de las manos y sindactilia de los dedos de los pies. Junto a leve dismorfia facial. Aunque no hay ningún miembro afectado de atresia de esófago/duodeno, creemos que cumplen criterios suficientes de síndrome de Feingold.
El motivo de presentar esta comunicación es dar importancia a la exploración física dismorfológica, como base en el reconocimiento de entidades sindrómicas, que como en estos casos es de gran importancia para el consejo genético.
Nueva deleción subtelomérica en 4p
L. Rodríguez 1, S. Climent 2, E. Mansilla 1, F. López-Grondona 1, M.L. Martínez-Fernández 1, M. Zollino 3 y M.L. Martínez-Frías 4
1Estudio Colaborativo Español de Malformaciones Congénitas (ECEMC) del Centro de Investigación sobre Anomalías Congénitas (CIAC). Instituto de Salud Carlos III. Ministerio de Sanidad y Consumo. Madrid. 2Departamento de Pediatría del Hospital de Ontinyent. Valencia. España. 3Istituto di Genetica Medica. Universitá Católica Sacro Cuore. Rome. Italy. 4Departamento de Farmacología. Facultad de Medicina. Universidad Complutense. Madrid. España.
El síndrome de Wolf-Hirschhorn (WHS), es un síndrome de genes contiguos ampliamente conocido cuya región crítica (WHSCR) se ha descrito localizada en el locus D4F26 en la banda 4p16.3. No obstante, con las nuevas técnicas moleculares, se han descrito varios casos con algunas manifestaciones clínicas del WHS, con una microdeleción que no involucra la WHSCR sino una región próxima. Esto sugiere que el locus D4F26 no sea la región crítica del WHS.
Presentamos el caso de una niña que nos fue remitida a los 4 años de edad, con peso, talla y PC por debajo del P3. A la exploración presenta rasgos dismórficos con frente amplia, nariz larga con puente nasal prominente y columela ancha, filtrum corto con labio superior evertido y orejas ligeramente rotadas hacia atrás con hélix borrado. Los genitales son normales. Habla monosílabos y entiende órdenes simples.
El cariotipo de alta resolución fue normal (850 bandas), y la aplicación de técnicas de hibridación in situ con fluorescencia (FISH) con el kit Multiprobe-T mostró ausencia de señal subtelomérica en uno de los cromosomas del par 4. La aplicación FISH con la sonda específica para WHSCR, mostró que no existía deleción de esa región. El cariotipo fue por tanto: 46,XX,del(4p).ish (tel4p-/tel4q +).ish 4p16.3(WHSCRx2) de novo.
Recientemente, Zollino et al (2003) han descrito un caso con algunas manifestaciones clínicas del WHS, con una deleción subtelomérica de 1,9 Mb en 4p16.3 conservando WHSCR, y proponen una nueva región crítica del WHS (WHSCR-2). Estos autores han encontrado la misma deleción de 1,9 Mb en nuestra paciente, lo que apoya su propuesta previa de que la región crítica del WHS es la WHSCR-2 (Zollino et al., 2003).
Consideramos que en pacientes en los que se sospeche un WHS, y no se detecte la microdeleción de la WHSCR se debería analizar la región subtelomérica 4p para descartar otras deleciones crípticas.
Este trabajo forma parte del proyecto PI020028 que ha sido financiado por el Fondo de Investigaciones Sanitarias (FIS), Instituto de Salud Carlos III. Ministerio de Sanidad y Consumo.
Caracterización de la trisomía parcial 9p23-p24 mediante técnicas de citogenética molecular en un niño con dismorfias y cariotipo 46,XY,add 16p1
M. Santos 1, A. Meneses 2, I. Gómez-Terreros 2 y C. Fuster 1
1Unitat de Biologia. Facultat de Medicina. Universitat Autònoma de Barcelona. 2Unidad de Dismorfología. Hospital Virgen del Rocío. Sevilla. España.
Introducción: Las modernas técnicas moleculares, en especial la hibridación genómica comparada de alta resolución (HR-CGH) y la hibridación in situ fluorescente (FISH) multicolor de 24 colores, se están mostrando herramientas muy potentes en la identificación de anomalías cromosómicas asociadas a malformaciones congénitas acompañadas o no de retraso mental.
Objetivo: Identificar el origen del material cromosómico adicionado al brazo corto del cromosoma 16 presente en el cariotipo de un niño de 2 años con estrabismo divergente de ojo derecho, dientes pequeños, boca de carpa, hipertelorismo con fisuras palpebrales antimongoloides, pectus excavatum, mancha acrómica en el cuello, pies zambos bilaterales, RGE e infecciones bronquiales frecuentes, bronquio supernumerario con estenosis traqueal, dificultades motoras finas y estudio cardiológico normal.
Resultados: Los perfiles de HR-CGH evidencian de forma clara la ganancia de material cromosómico correspondiente a la región 9p23-pter. La aplicación posterior de la técnica de FISH ha permitido confirmar la presencia de material del cromosoma 9 adicionado en 16p.
Discusión: Mientras el síndrome de la trisomía 9p es relativamente frecuente y se caracteriza por la presencia anomalías craneofaciales asociadas a la duplicación de la banda 9p22, la duplicación del extremo terminal 9p es rara. Nuestro paciente es el cuarto caso descrito en la literatura médica con trisomía 9p23-p24. En los otros 2 casos el fragmento duplicado se localizaba en el mismo brazo cromosómico 9p mientras que en el nuestro se encuentra translocado al 16p. La comparación de datos clínicos realizada evidencia la presencia de hipertelorismo y fisuras palpebrales antimongoloides en los 3 pacientes.
Agradecimientos: Financiación recibida por SAF (2003-03894) y CIRIT (2001, SGR-00201). Beca de la Generalitat de Catalunya (2002FI 00281).
Anomalías cromosómicas subteloméricas en pacientes con retraso mental idiopático (RMI): estudio comparativo entre hibridaciónin situcon fluorescencia (FISH) ymultiplex ligation-dependent probe amplification (MLPA)
A. Delicado 1, M. Palomares 1, P. Lapunzina 1, D. Arjona 2, C. Amiñoso 2, L. Fernández e I. López Pajares 1
1Servicio de Genética Médica. 2Unidad de Secuenciación. Hospital Universitario La Paz. Madrid. España.
Introducción: El retraso mental es un trastorno común que afecta al 1-3 % de la población general. Su etiología permanece sin identificar en el 50 % de los pacientes. En los últimos 5 años numerosos estudios han demostrado que las reestructuraciones teloméricas en desequilibrio son una causa importante de retraso mental con una frecuencia de entre 3-12 %.
Material y métodos: Se estudian 45 pacientes con retraso mental de origen no aclarado asociado a rasgos dismórficos, malformaciones y/o historia familiar de retraso mental. Los estudios iniciales se realizaron mediante FISH con sondas múltiples subteloméricas (Chromoprobe Multiprobe-T system, Cytocell Technologies Ltd.) y posteriormente con MLPA (Kit Salsa PO-36-telomers, MRC-Holland).
Resultados: Con FISH se identificaron 5 pacientes con anomalías subteloméricas en desequilibrio (11 %). Con MLPA se obtuvieron resultados similares a excepción del paciente 5 en el cual la pérdida de 15q no fue detectada (tabla 1).

Conclusiones: 1. La frecuencia de anomalías subteloméricas (11 %) es importante y justifica la aplicación de dicha metodología en pacientes con RM idiopático. 2. El FISH es un método laborioso y caro. Sin embargo, MLPA es una técnica exacta, rápida y menos costosa que el FISH. Por tanto, constituye el método de elección para detectar reestructuraciones subteloméricas en una población de pacientes con RM idiopático, cuando la técnica sea ampliamente validada. 3. FISH con las sondas específicas debe utilizarse para reafirmar el diagnóstico.
Duplicación hipofisaria con maldesarrollo frontonasal y del SNC
E. González-Obeso, C. Morales, A. Mariño, E. García-Fernández y J.I. Rodríguez
Departamento de Anatomía Patológica. Hospital Universitario La Paz. Madrid. España.
La duplicación hipofisaria es una malformación rara que puede aparecer aislada o asociada a distintas malformaciones craneofaciales. Hay alrededor de 24 casos descritos desde 1880.
Se presenta el octavo caso de autopsia, una niña (46,XX) de 2 meses y 20 días de edad con malformación frontonasal (hipertelorismo, raíz nasal ancha, punta nasal bífida y cráneo anterior bífido), macrostomía, retrognatia, lengua bífida, fusión ósea maxilomandibular y fusión de la lengua al paladar y al labio inferior por un pólipo velloso oral, ambas fusiones corregidas quirúrgicamente.
La silla turca estaba ensanchada, tenía apófisis clinoides poco evidentes y dos cavidades que albergaban dos hipófisis completas con dos infundíbulos. Las tres fosas craneales estaban estrechadas en sentido anteroposterior. El sistema nervioso central (SNC) presentaba agenesia de bulbos olfatorios y de cuerpo calloso, morfología anómala de tronco y médula espinal y heterotopias de sustancia gris en sustancia blanca occipital y glial en meninge.
Las malformaciones frontonasales descritas en los 7 casos autópsicos son, por orden de frecuencia, hipertelorismo (6 casos), masa intraoral (epignatus) (4), lengua bífida (3), duplicación del surco anterior de la médula espinal (1) y línea de inserción capilar en la frente en forma de "V" (1). Las malformaciones del SNC encontradas fueron duplicación hipofisaria, ensanchamiento de la silla turca y anomalías hipotalámicas (7), agenesia de cuerpo calloso (5), agenesia de bulbos olfatorios (2) y heterotopias gliales y/o neuronales (1). El caso que se presenta tiene mayor similitud con el descrito por Hori (1983), que, además, tenía paladar hendido, anomalías en el polígono de Willis, extensión de la duplicidad del surco anterior de la médula espinal hasta la columna torácica pero no tenía agenesia de bulbos olfatorios.
Existen varias hipótesis acerca del origen de la duplicidad hipofisaria y las malformaciones que puede asociar, sin embargo, ninguna es aún definitiva.
El MLPA como método diagnóstico de elevada eficiencia en el síndrome de microdeleción 22q11.2
I. López Pajares 1, L. Fernández 1, P. Lapunzina 1, D. Arjona 2, D. Elorza 3, L. García-Guereta 4, M.L. de Torres 1, M.A. Mori 1, M. Palomares 1, M. Burgueros 4, J. Pérez 3 y A. Delicado 1
1Servicio de Genética Médica. 2Unidad de Secuenciación. Servicios de 3Neonatología y 4Cardiología Infantil. Hospital Universitario La Paz. Madrid. España.
El síndrome de microdeleción 22q11.2 se diagnostica normalmente por hibridación in situ de fluorescencia (FISH) convencional, siendo generalmente determinados los puntos de rotura cromosómicos y el tamaño de la deleción mediante tests de segregación de microsatélites o hibridaciones más específicas. El multiplex ligation-dependent probe amplification (MLPA) puede ser un recurso útil para detectar haploinsuficiencia en 22q11.2 y en otras regiones cromosómicas asociadas al síndrome de DiGeorge/velocardiofacial en un único ensayo. Hemos comparado los resultados de las tres técnicas en una serie de 30 pacientes afectados del síndrome de deleción 22q11.2 y previamente diagnosticados por FISH. El MLPA diagnosticó satisfactoriamente a todos los pacientes, y con respecto al tamaño de la deleción, los resultados de la MLPA coincidieron con los tests de microsatélites para todos los pacientes, y resolvieron 7 casos que eran no informativos por microsatélites debido a fallos en la amplificación, a marcadores homozigotos o a la ausencia de datos parentales. Estos hallazgos apoyan la utilidad del MLPA como técnica de diagnóstico para el síndrome de microdeleción 22q11.2 como un método rápido, sencillo eficiente y fácilmente reproducible.
Síndrome de Simpson-Golabi-Behmel. Una nueva familia con una mutación intrónica
L.F. Magano 1, R. Gracia 1, M. Segovia 2, R.M. Valdez 2, I. Incera 1, P. Arias 1 y P. Lapunzina 1
1Hospital Universitario La Paz. Madrid. España. 2Instituto Nacional de Genetica. Buenos Aires. Argentina.
El síndrome de Simpson-Golabi-Behmel (SGBS) es un síndrome ligado al cromosoma X caracterizado por un sobrecrecimiento prenatal y posnatal (gigantismo) que incluyen macroglosia, macrosomía, anormalidades renales y esqueléticas y un riesgo elevado de tumores embrionarios, como el de Wilms y el de hígado.
Las deleciones y/o mutaciones que implican el gen glipican-3 (GPC3) en Xq26 están asociadas al síndrome (SGBS). El gen GPC3 codifica para una proteína de la familia de los proteoglucanos, glipican-3, que se supone desempeña un papel importante en control del crecimiento en los tejidos mesodérmicos embrionarios, en los cuales se expresa selectivamente. Parece formar un complejo con el IGF-2, pudiendo modular la acción de éste.
Estudiamos el gen GPC3 en una familia con SGBS. El caso índice y su tío materno presentan una mutación intrónica (IVS2 + 1 G > A) que predecimos produce una pérdida de función de la proteína glipican-3.
Alteraciones cromosómicas finas en translocaciones aparentemente balanceadas: un aspecto a tener en cuenta en el asesoramiento genético
M.L. Martínez-Fernández 1, L. Rodríguez 1, F. López-Grondona 1, E. Mansilla 1, E. Cruz 2, C. Sánchez 2, N. Tommerup 3 y M.L. Martínez-Frías 1,4
1Centro de Investigación de Anomalías Congénitas (CIAC). Instituto de Salud Carlos III. Ministerio de Sanidad y Consumo. Madrid. 2Servicio de Pediatría. Hospital Santa Bárbara. Puertollano. Ciudad Real. España. 3Instituto Panum. Copenhague. Dinamarca. 4Departamento de Farmacología. Facultad de Medicina. Universidad Complutense de Madrid. España.
En la actualidad, gracias a las técnicas de citogenética de alta resolución (550-850 bandas G) se están detectando alteraciones cada vez más finas en niños con defectos congénitos. Cuando la alteración es una translocación "aparentemente balanceada" y el niño tiene alguna anomalía congénita, cabe pensar que dicha alteración no sea realmente balanceada, ya que existe la posibilidad de que se hayan producido anomalías durante los reordenamientos del apareamiento y la recombinación meiótica. Para determinarlo es conveniente realizar, al menos, un cariotipo de alta resolución.
Presentamos el caso de un niño de 6 meses, que llega a nuestro centro con el diagnóstico citogenético de translocación, aparentemente balanceada, entre los cromosomas 2 y 12. Dicha alteración se había diagnosticado por amniocentesis ya que 2 hermanos de la madre y la abuela tenían una translocación balanceada entre los cromosomas 11 y 12. El niño presentaba dismorfias craneofaciales y retraso psicomotor, por lo que nos fue remitido para estudio cromosómico de alta resolución. En dicho estudio se confirmó la translocación pero, además, se identificó una deleción del cromosoma 12 involucrado en la translocación, que fue confirmada mediante estudio de hibridación genómica comparada (CGH). El niño era, por tanto, portador de una translocación no balanceada entre los cromosomas 2 y 12.
Este caso es un claro ejemplo de que en pacientes con defectos congénitos graves o leves que sean portadores de una alteración cromosómica aparentemente balanceada y con cariotipo de baja resolución, no se debería descartar la posibilidad de que exista un desbalance que no se haya identificado, a pesar de que uno de los padres sea portador aparentemente de la misma alteración cromosómica. Esta posibilidad hay que tenerla presente para ofrecer un adecuado asesoramiento a la familia.
Disomía uniparental materna del cromosoma 14 en un paciente portador de una translocación robertsoniana 13;14
M.A. Mori 1, A. Delicado 1, P. Lapunzina 1, M.L. de Torres 1, M.C. Roche 2, J. Arcas 2 e I. López-Pajares 1
Servicios de 1Genética Médica y 2Neurología Pediátrica. Hospital Universitario La Paz. Madrid. España.
La disomía uniparental (DU) se produce al heredar ambos cromosomas homólogos del mismo progenitor. La DU ha sido descrita en prácticamente todos los cromosomas, estando asociada, en alguno de ellos, a una alteración en el fenotipo debido al fenómeno conocido como imprinting genómico.
Los portadores de anomalías cromosómicas estructurales, incluidas las translocaciones robertsonianas, tienen un riesgo aumentado para DU.
El propósito es un niño de 7 meses de edad procedente de la consulta de Neurología Infantil. Presentaba hipotonía, retraso motor y rasgos dismórficos: dolicocefalia, nariz respingona, frente amplia, boca en "V" invertida, filtrum corto, cara redonda, facies peculiares y pliegue palmar único.
El estudio cromosómico reveló que el paciente era portador de una translocación robertsoniana 13;14 de origen materno. El análisis molecular utilizando 5 microsatélites del cromosoma 14 desveló la presencia de una heterodisomía de origen materno de dicho cromosoma.
El presente caso demuestra la importancia de descartar una DU en aquellos casos de portadores de una translocación robertsoniana asociados a rasgos dismórficos. Asimismo en caso de detección prenatal de portadores de translocaciones robertsonianas debe considerarse la posibilidad de realizar estudios de disomía puesto que el riesgo que esta se produzca es de aproximadamente 0,5 %.
Síndrome de Stickler: caso familiar
S. Climent Alberola 1, I. Belda Galiana 1, P. Correcher Medina 2, I. Victoria Miñana 2 y F. Calvo Rigual 2
1Unidad de Pediatría. Hospital General de Ontinyent. 2Servicio de Pediatría. Hospital Lluís Alcanyís. Xàtiva. Valencia. España.
Presentamos el caso de 2 hermanos, niño y niña de 7 años y 20 meses, respectivamente.
Nos es remitida la niña por rasgos dismórficos. La exploración física muestra un adecuado desarrollo pondoestatural, facies con ojos prominentes, escleróticas azuladas, pliegues epicánticos, puente nasal ancho y deprimido, retrognatia, úvula bífida y retraso en la fluxión dental. Extremidades: dedos largos e hiperextensibilidad. Desarrollo psicomotor normal. Se practica serie esquelética que se informa como normal y exploración audiométrica dentro de la normalidad.
Al realizar la historia clínica, se sabe que su hermano ha presentado dos desprendimientos de retina en abril de 2004 y enero de 2005, así como que presentó fisura palatina al nacimiento. A la exploración presenta ojos prominentes (miopía magna > 15 dioptrías), pliegues epicánticos, puente nasal ancho, cara aplanada con mandíbula pequeña, fisura palatina intervenida. Extremidades con dedos largos, hiperextensibilidad y engrosamiento en articulaciones interfalángicas, codos y rodillas. Desarrollo psicomotor normal. Estudio ecocardiográfico normal. Pendiente estudio óseo.
Conocidos los datos descritos se realiza estudio oftalmológico a la niña, objetivándose una miopía grave.
El síndrome de Stickler, en sus dos formas tipo 1 (75 %) y tipo 2 (25 %), se hereda de forma autosómica dominante con una expresividad muy variable, tanto interfamiliar como intrafamiliar. Alrededor del 70 % de los casos se debe a mutaciones del gen COL2A1 (tipo 1) y COL11A1 (tipo 2). Se ha descrito también una forma no ocular (OSMED) COL11A2.
Presentamos este caso familiar como referente a tener en cuenta ante hallazgos muy frecuentes asociados a esta entidad, como la secuencia de Pierre-Robin, en los cuales habrá que realizar un adecuado seguimiento y diagnóstico.
Displasia ectodérmica hipohidrótica. A propósito de dos hermanos
J. Guerrero Fernández, M.ªA. Molina Rodríguez, M.T. García Ascaso y R. Gracia Bouthelier
Servicio de Endocrinología Pediátrica. Hospital Infantil La Paz. Madrid. España.
Introducción: La displasia ectodérmica hipohidrótica ligada al cromosoma X es una enfermedad que afecta a varones y caracterizada por hipoanhidrosis o anhidrosis, hipodoncia e hipotricosis.
Casuística: El primer caso, ecuatoriano y de 13 años de edad, consultó por hipotricosis generalizada e hipodoncia. Entre los antecedentes personales destacaban varios ingresos por golpes de calor, además de estudios diagnósticos por dicho motivo que resultaron normales. El hermano, de 10 años, presentaba las mismas características así como frecuentes antecedentes de episodios febriles sin foco.
El diagnóstico, en ambos casos, se hizo al demostrar la ausencia de varios alvéolos dentarios en la radiografía y la escasa respuesta sudorípara a la inyección subcutánea de pilocarpina. El estudio inmunológico permitió descartar déficit inmunológicos.
Discusión: La displasia ectodérmica hipohidrótica ligada al cromosoma X (Xq12-13; MIM: 305100) determina alteraciones de estructuras ectodérmicas, fundamentalmente pelo, piel, uñas y dientes. Tales defectos determinan hipotricosis, que resulta más llamativa en cejas y cuero cabelludo, alteraciones dentarias (ausencia, dientes cónicos, etc.), de uñas y aplasia o hipoplasia de glándulas sudoríparas ecrinas con la consiguiente reducción de la sudoración y trastorno en la capacidad para controlar la pérdida de calor haciendo a estos niños especialmente susceptibles al sobrecalentamiento ambiental.
Una forma distinta de displasia ectodérmica hipohidrótica ligada a cromosoma X ha sido descrita en asociación con inmunodeficiencia humoral y, consecuentemente, con un peor pronóstico. Dado que la presentación fenotípica es similar en ambos procesos, siempre será preciso un estudio inmunológico que permita establecer el riesgo a infecciones bacterianas potencialmente graves. En base a esto, el interés de ambos casos se debe, de una parte, a la necesidad de incluir esta entidad dentro del diagnóstico diferencial del síndrome febril recidivante, y, de otra, a que el estudio inmunitario de estos pacientes condiciona el valor que ha de darse a cada proceso febril que padezcan: golpe de calor frente a infección potencialmente grave.
Microtia. Una malformación congénita de alta frecuencia en los Andes
L. Ortiz y M. Jijón
Departamento de Docencia e Investigaciones. Servicio de Genética médica. Hospital de Niños Baca Ortiz. Quito. Ecuador.
Presentamos en el siguiente trabajo los casos de microtia atendidos en la consulta externa de Genética Médica del Hospital de Niños Baca Ortiz de Quito (Ecuador), y que ocurrieron en el período comprendido entre enero de 1999 a diciembre de 2004.
En 1983 el Estudio Colaborativo Latinoamericano de Malformaciones Congénitas (ECLAMC) afirma que [...] "la incidencia de microtia en dos hospitales de Quito, era cinco veces mayor (16/10.000 nv) que el resto de los hospitales de la red ECLAMC (3.2/10.000) distribuidos en 24 ciudades de otros 6 países sudamericanos. La microtia está relacionada a las regiones de gran altura sobre el nivel del mar, pero su etiología aún no establecida sugiere una causal multifactorial".
Nuestra casuística comprende 187 casos, cifra muy alta comparada con otros comunicados por nosotros identificados. Presentamos la casuística distribuida por sexo, edad, lado afectado, sea unilateral o bilateral, agenesias, grados de presentación. En todos los casos se hizo valoración de audición, radiografía de columna vertebral y ecografía renal. Distinguimos los casos aislados de los sindrómicos y aquellos que se presentaron con repetición en la misma familia.
Síndrome de Langer-Giedion con t(17;19)(q12;p13)pat
A. Pardo, T. Aravena, G. Franco y S. Castillo Taucher
Sección Genética. Hospital Sótero del Río. Hospital Clínico Universidad de Chile. Santiago. Chile.
Probando de 3 años 7 meses, segunda hija de pareja joven, sana, no consanguínea. Hermana sana. Gestación y parto normales, peso nacimiento 3 250 g (P50), talla 48 cm (P50) y CC 33,5 cm (P50). Derivada a genética 3 meses 24 días por dismorfias faciales (dolicocefalia, hipertelorismo ocular, nariz bulbosa con columela corta, filtrum largo, boca grande con labio superior fino, micrognatia, orejas bajas) y braquifalangia, uñas displásicas y fetal pads. Regresa 2 años y 5 meses, peso 13,4 kg (P25), talla 94 cm (P50) y CC 46 cm (P50), por aparición de exostosis en ambas escápulas, fémur y tibia derechos, y peroné izquierdo.
Cariogramas: niña: 46,XX, t(17;19)(q12;p13)pat, madre: normal y padre: 46,XY, t(17;19)(q12;p13). Radiografías: exostosis. TC cerebral normal. Diagnóstico síndrome de Langer-Giedion. Faltan examen físico y radiológico del padre, y cariotipo en hermana y/u otros familiares paternos.
Caso sin diagnóstico # 1
I. Bueno y F.J. Ramos
Hospital Clínico Universitario. Facultad de Medicina. Universidad de Zaragoza. España.
Paciente varón con desproporción craneofacial, facies "fetal", cardiopatía congénita (PDA), atresia de coanas, coloboma bilateral, criptorquidia, anomalías vertebrales y costales, hidrocefalia y retraso mental.
Caso sin diagnóstico # 2
I. Bueno y F.J. Ramos
Hospital Clínico Universitario. Facultad de Medicina. Universidad de Zaragoza. España.
Paciente varón de 12 años con talla baja, retraso mental, cardiopatía congénita (CIV), frente amplia, hipertelorismo ocular, estrabismo, nariz corta, filtrum alargado, paladar elevado y braquidactilia. Madre con fenotipo craneofacial similar e inteligencia borderline.
