




La trombocitopenia inmune primaria (PTI) es poco frecuente en la infancia, pero es la causa más habitual de trombocitopenia. Se han intentado establecer factores de riesgo para predecir su evolución, con el objetivo de poder optimizar su manejo, que se ha modificado en los últimos años, debido, entre otros factores, a una atención más especializada.
Material y métodosEstudio retrospectivo, observacional y analítico de los pacientes con PTI, en un periodo de 3 años, en una consulta especializada en Hematología Pediátrica.
ResultadosDesde el punto de vista epidemiológico, clínico y analítico, las características de esta serie son similares a las de otros grupos. La mayoría de los pacientes (23/31; 74,2%) presentaron una PTI de duración menor de 12 meses, sin complicaciones graves relacionadas con la enfermedad ni con el tratamiento. Se establecieron como factores de riesgo relacionados con una evolución tórpida (supervivencia libre de eventos [SLE] menor), sin alcanzar la significación estadística, el sexo femenino, la edad mayor de 10 años, la leucopenia, la ausencia de trombocitopenia grave inicial y la atención no especializada. La ausencia de antecedente de infección se relacionó significativamente con una SLE menor.
ConclusionesLos factores de riesgo de evolución tórpida de PTI epidemiológicos y analíticos de este estudio coinciden con los descritos en la literatura. Presentaron una SLE menor los pacientes tratados antes del inicio de la atención especializada. Estos datos parecen apoyar la recomendación actual de que las enfermedades poco frecuentes, como esta, se controlen en unidades especializadas.
Although primary immune thrombocytopenia (ITP) is rare in childhood, it is the most frequent cause of thrombocytopenia. There have been attempts to establish risk factors to predict the progression of the disease in order to optimise its management, which has changed in recent years due to, among other reasons, specialised care.
Material and methodsA retrospective, observational and analytical study was conducted on patients diagnosed with ITP over a 3-year period in a Paediatric Haematology specialist clinic.
ResultsFrom the epidemiological, clinical and analytical point of view, the characteristics of this group are similar to others. Most of the patients (23/31, 74.2%) had ITP for less than 12 months, with there being no serious complications related to the disease or the treatment received. It was established that risk factors were related to being slowly evolving (lower event-free survival (EFS)) with no statistical significance, female gender, age over 10 years, leukopenia absence of initial severe thrombocytopenia, and non-specialised care. The absence of a history of infection was significantly related to a lower EFS.
ConclusionsThe epidemiological and analytical risk factors for a slowly evolving ITP are the same that described in the literature. Patients treated before the beginning of specialised care also had a lower EFS. These data seem to support the current recommendation that rare diseases should be managed in specialised units.
La trombocitopenia inmune primaria (PTI) es una enfermedad caracterizada por una disminución aislada de la cifra de plaquetas por debajo de 100.000/mm3, sin encontrar una causa desencadenante que la explique. Actualmente se establece el término «crónica» a partir de 12 meses de evolución desde el diagnóstico1,2.
Es la causa más frecuente de trombocitopenia de presentación aguda en un niño sano, pero no es una enfermedad frecuente. La incidencia en menores de 15 años se estima en unos 5 casos por 100.000 habitantes/año3.
Se caracteriza fundamentalmente por sintomatología hemorrágica a 3 niveles: piel, mucosas y órganos internos. La mayoría de los pacientes están asintomáticos o tienen afectación cutáneo-mucosa. Se han desarrollado diferentes escalas clínicas para medir de forma objetiva la intensidad de los sangrados, siendo una de las más empleadas la desarrollada por la OMS: grado 0, sin evidencia de sangrado; grado 1: sangrados leves (petequias, equimosis, mucosas, sangrado retiniano sin alteraciones visuales); grado 2: sangrados mayores (melena, hematemesis, hematuria, hemoptisis); grado 3: cualquier sangrado que requiera transfusión de hematíes; grado 4: sangrados retinianos con alteraciones en la visión, sangrados en sistema nervioso central. Solamente el 3% de los pacientes pediátricos presentan sintomatología de sangrado clínicamente significativo, siendo las manifestaciones más frecuentes la epistaxis y el sangrado gastrointestinal3. Las hemorragias parecen estar relacionadas con la gravedad de la trombocitopenia, aunque no existe una correlación exacta ni directa entre estas variables. La complicación más grave es la hemorragia intracraneal (HIC), cuya frecuencia oscila entre el 0,1 y el 0,6%. No se ha establecido un claro factor predictor del riesgo de HIC en estos pacientes3-6.
El diagnóstico de PTI es de exclusión. Una historia personal y familiar y un examen físico normales (salvo las manifestaciones hemorrágicas), asociados a la presencia de trombocitopenia aislada, constituyen las herramientas clave para establecer el diagnóstico. La mayoría de los pacientes presentan antecedentes de infección viral (60% infección de vías altas y exantemas), inmunización (más frecuente en relación con la vacuna triple vírica) o infección bacteriana en las semanas previas3,4. Cualquier otra alteración analítica que no sea la trombocitopenia aislada, y/o anomalías en la exploración física (dismorfias, visceromegalias, etc.) o en la historia clínica (infecciones de repetición, fiebre, medicamentos, etc.) deben, al menos, hacer replantearse el diagnóstico de PTI7,8.
La PTI es una enfermedad benigna y autolimitada. Diversos registros y publicaciones han demostrado una remisión superior al 60% en los 6 meses tras el diagnóstico, independientemente del tratamiento recibido3,9. La identificación de factores predictores de la remisión es difícil de establecer; se ha encontrado una relación entre la evolución favorable y los siguientes: sexo masculino, edad<10 años, inicio abrupto, presencia de infección previa y recuento de plaquetas muy bajo (<5.000/mm3)10-13. También la presencia de leucopenia parece asociarse a un mayor riesgo de PTI persistente o crónica14.
Los fármacos más frecuentemente empleados para su tratamiento han sido los corticoides y las inmunoglobulinas intravenosas (IGIV)15-18. Clásicamente la indicación de tratamiento se basaba en aumentar las cifras de plaquetas. Con los datos que tenemos en la actualidad, sobre la rareza del sangrado significativo, la ausencia de evidencia de que el tratamiento prevenga el sangrado grave, y los costes y toxicidades conocidos de los diferentes tratamientos, se aboga por un manejo más conservador, recomendando la observación para los niños asintomáticos o mínimamente sintomáticos. En el caso de recibir tratamiento, al no existir uno que haya demostrado su superioridad, es recomendable valorar los pros y contras con los pacientes y sus familias para alcanzar un plan individualizado3,9,19. Este cambio de estrategia terapéutica queda recogido en los diferentes protocolos y guías publicados en los últimos años, como el protocolo de la Sociedad Española de Hematología y Oncología Pediátricas (SEHOP)19-21.
En 2013 se puso en marcha una consulta especializada en Hematología Pediátrica en el Hospital Infantil Miguel Servet de Zaragoza, para encargarse del diagnóstico, tratamiento y seguimiento de los pacientes diagnosticados de procesos hematológicos no oncológicos, entre ellos, los pacientes con PTI. Hasta esa fecha, los casos de PTI en este centro eran tratados por diferentes profesionales no especialistas en Hematología Pediátrica. A partir de la puesta en marcha de la consulta especializada, se emplearon de forma sistemática protocolos avalados por sociedades científicas expertas en Hematología Pediátrica20.
El objetivo de este trabajo es conocer las características clínicas, analíticas y evolutivas de los casos valorados en esa consulta especializada, y estudiar los factores de riesgo que pudieron relacionarse con una evolución más tórpida.
Población y métodosSe realizó un estudio retrospectivo, observacional y analítico mediante la revisión de historias clínicas de pacientes diagnosticados de PTI, atendidos entre enero de 2013 y octubre de 2016 en la consulta especializada en Hematología Pediátrica en el Hospital Infantil Miguel Servet de Zaragoza.
Se incluyeron los pacientes menores de 15 años diagnosticados de PTI, atendidos en esta consulta en ese intervalo de tiempo, independientemente de la fecha del diagnóstico inicial. Se excluyeron del estudio los pacientes sin datos suficientes de seguimiento.
Se emplearon los criterios diagnósticos, de respuesta y los tratamientos recogidos en el protocolo sobre PTI de la SEHOP vigente20.
Los datos recogidos incluían variables demográficas y epidemiológicas, analíticas, de tratamiento, respuesta al mismo e información sobre el seguimiento y la evolución. Toda la información fue recogida bajo el cumplimiento de la Ley de Protección de Datos vigente.
Las variables se compararon mediante las pruebas de χy y t de Student. Para establecer relaciones entre las variables se empleó el estudio de regresión logística binaria, siendo la variable dependiente el tipo de PTI según el tiempo de evolución (menor o igual a 12 meses/mayor de 12 meses). Se realizó también un estudio de supervivencia, definiendo supervivencia libre de eventos (SLE) como el tiempo transcurrido en meses desde el diagnóstico hasta la aparición de (lo que ocurriese primero): descenso de plaquetas <30.000/mm3, muerte, alta, o final del estudio (31 de octubre de 2016). Las curvas de supervivencia se realizaron mediante el método de Kaplan-Meier. Se comparó la diferencia en la SLE en función de diferentes factores clínicos, diagnósticos y terapéuticos (edad, sexo, presencia de sangrado mucoso, antecedente infeccioso, cifra de plaquetas y de leucocitos, atención en consulta especializada o previa a la puesta en marcha de la misma) mediante análisis univariable por Kaplan-Meier, empleando el test log-rank. La significación estadística se estableció para un valor de p o B menor de 0,05. El análisis estadístico se realizó utilizando el programa IBM SPSS versión 24.
ResultadosEntre enero de 2013 y octubre de 2016 fueron valorados en la consulta de Hematología Pediátrica 509 pacientes por algún trastorno hematológico benigno. De estos, 44 (8,6%) estaban diagnosticados de trombocitopenia, y 31 (70% de las trombocitopenias) tenían un diagnóstico de PTI. Con estos datos y los demográficos del Instituto Aragonés de Estadística se puede establecer una incidencia en menores de 15 años de 2,6 casos por 100.000 habitantes/año.
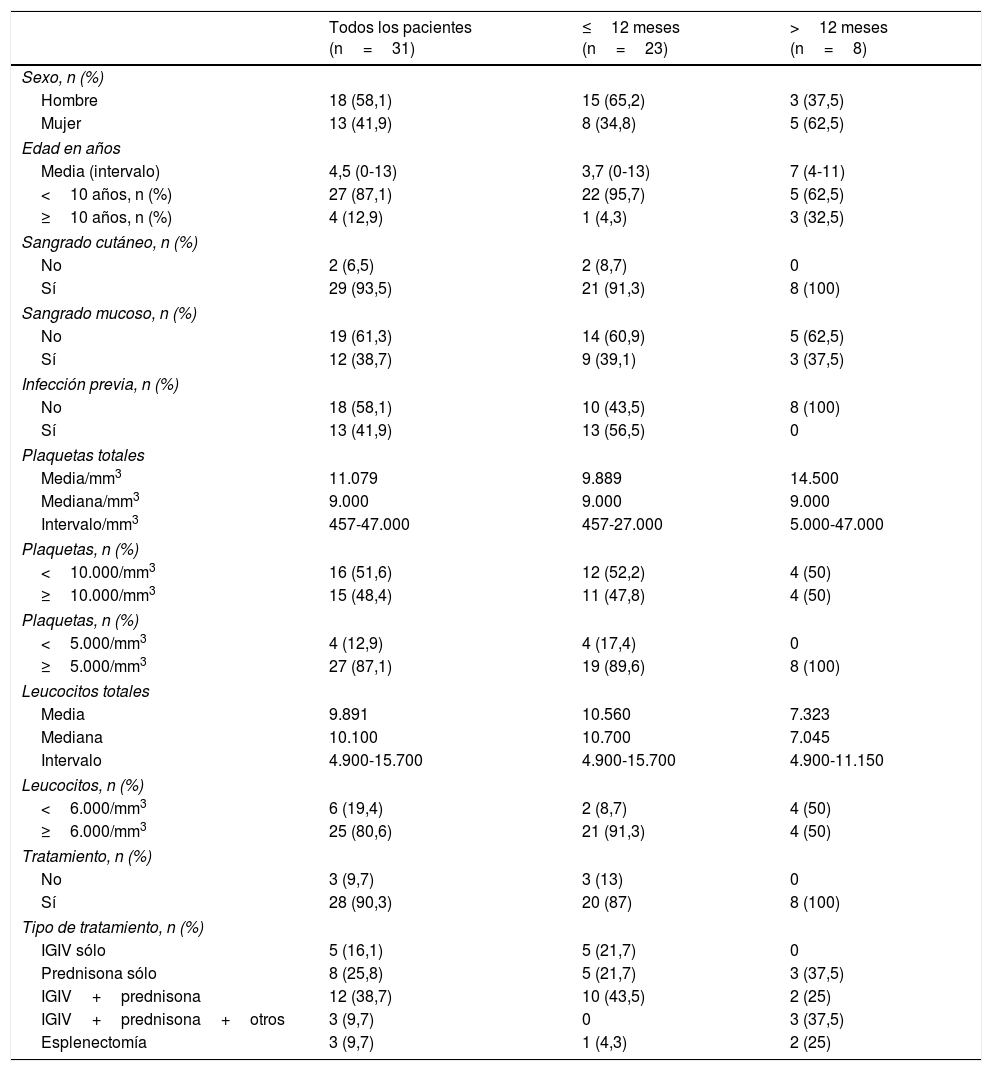
La distribución por género fue de 18 hombres frente a 13 mujeres (relación 1,3:1), con una edad media al diagnóstico de 4,5 años, mediana de 4 años y un intervalo de 0-13 años. La mayoría de los pacientes (27/31) tenían una edad menor de 10 años al diagnóstico.
En relación con las manifestaciones clínicas, 2 pacientes (6,5%) no presentaban ningún signo de sangrado, estableciéndose el diagnóstico al hacer una analítica por otros motivos. En los 29 restantes se objetivaron signos de sangrado: cutáneo en todos ellos, y mucoso en 12 casos (38,7%), siendo la epistaxis el sangrado más habitual (10/12; 83%), seguido del sangrado gastrointestinal. En ningún paciente se registró el antecedente de vacunación, existiendo antecedente infeccioso previo en 13 casos (41,9%).
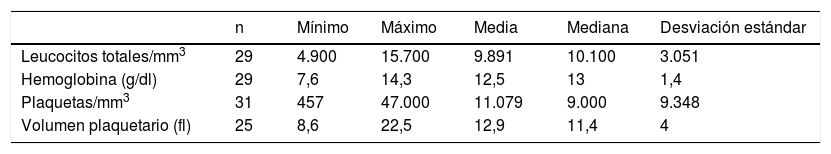
La cifra media de plaquetas al diagnóstico fue de 11.000/mm3, y la mediana de 9.000/mm3. El 83,8% de pacientes presentaban <20.000 plaquetas/mm3 al diagnóstico y se encontró trombocitopenia muy grave (<5.000 plaquetas/mm3) en un 13% de la muestra. Las plaquetas al diagnóstico de la serie no superaron en ningún caso las 50.000/mm3. Las cifras de hemoglobina iniciales fueron anormales en 2 casos, en los que se realizó un estudio de médula ósea, que confirmó el origen periférico de la trombocitopenia. Seis pacientes (19,4%) presentaron leucocitos <6.000/mm3 al diagnóstico, y de estos, la mayoría (66%) evolucionaron hacia PTI crónica (tabla 1).
De los 31 pacientes, 3 (9,7%) no recibieron ningún tratamiento médico a lo largo del seguimiento; todos ellos evolucionaron a la curación en los 6 meses siguientes. Trece (46,8%) recibieron un tratamiento, siendo el más empleado la prednisona (8 pacientes), seguido de las IGIV (5 pacientes). Doce pacientes precisaron recibir 2 líneas de tratamiento, por escasa respuesta clínica y/o analítica, y en 3 casos se emplearon otras terapias (dexametasona, rituximab, análogos de la trombopoyetina, esplenectomía). Un paciente presentó epistaxis que precisó terapia transfusional por repercusión hemodinámica. No se registró ninguna muerte ni ningún sangrado amenazante para la vida en toda la muestra en el periodo de estudio. Según la escala de sangrados de la OMS, la mayoría de los pacientes (28/31 90,3%) presentaron hemorragia grados 1-2. Se registraron 2 casos con grado 0, un caso con grado 3 y no hubo ningún caso con sangrado grado 4.
Veintinueve pacientes (93,5%) alcanzaron una cifra >100.000 plaquetas/mm3, con un tiempo medio de 150 días y una mediana de 65 días. Once casos (35,4%) presentaron un descenso por debajo de 30.000 plaquetas/mm3 durante el seguimiento, con un intervalo medio de 87 días y una mediana de 30 días desde el diagnóstico. Todos los que disminuyeron las cifras de plaquetas recibieron, en algún momento al menos, un tratamiento: 3 pacientes solamente prednisona, 4 pacientes prednisona e inmunoglobulinas y 4 pacientes al menos 3 fármacos inmunomoduladores.
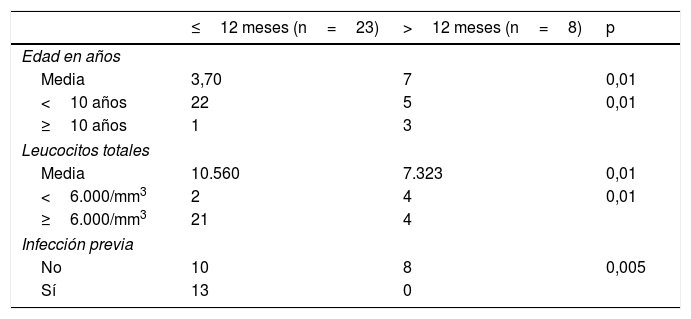
De los 31 pacientes, la mayoría (23/31; 74,2%) resolvieron la trombocitopenia en los siguientes 12 meses al diagnóstico, y 8 pacientes (25,8%) cumplieron criterios de PTI crónica. Los pacientes con PTI crónica eran más frecuentemente mujeres, con una edad media más elevada que la muestra total, una cifra media de plaquetas más elevada al diagnóstico, leucocitos más bajos al diagnóstico y sin antecedente de infección previa. En los pacientes con PTI de más de 12 meses de evolución, comparándolos con los pacientes con PTI aguda o recurrente de menos de 12 meses de evolución, se encontró una relación estadísticamente significativa con una edad más elevada al diagnóstico, la presencia de leucopenia y la ausencia de infección. No se encontró una relación estadísticamente significativa en relación con la cifra de plaquetas, la presencia de sangrado mucoso, ni el tratamiento recibido (tablas 2 y 3).
Características clínicas, analíticas y terapéuticas de los pacientes en función del tiempo de evolución
| Todos los pacientes (n=31) | ≤12 meses (n=23) | >12 meses (n=8) | |
|---|---|---|---|
| Sexo, n (%) | |||
| Hombre | 18 (58,1) | 15 (65,2) | 3 (37,5) |
| Mujer | 13 (41,9) | 8 (34,8) | 5 (62,5) |
| Edad en años | |||
| Media (intervalo) | 4,5 (0-13) | 3,7 (0-13) | 7 (4-11) |
| <10 años, n (%) | 27 (87,1) | 22 (95,7) | 5 (62,5) |
| ≥10 años, n (%) | 4 (12,9) | 1 (4,3) | 3 (32,5) |
| Sangrado cutáneo, n (%) | |||
| No | 2 (6,5) | 2 (8,7) | 0 |
| Sí | 29 (93,5) | 21 (91,3) | 8 (100) |
| Sangrado mucoso, n (%) | |||
| No | 19 (61,3) | 14 (60,9) | 5 (62,5) |
| Sí | 12 (38,7) | 9 (39,1) | 3 (37,5) |
| Infección previa, n (%) | |||
| No | 18 (58,1) | 10 (43,5) | 8 (100) |
| Sí | 13 (41,9) | 13 (56,5) | 0 |
| Plaquetas totales | |||
| Media/mm3 | 11.079 | 9.889 | 14.500 |
| Mediana/mm3 | 9.000 | 9.000 | 9.000 |
| Intervalo/mm3 | 457-47.000 | 457-27.000 | 5.000-47.000 |
| Plaquetas, n (%) | |||
| <10.000/mm3 | 16 (51,6) | 12 (52,2) | 4 (50) |
| ≥10.000/mm3 | 15 (48,4) | 11 (47,8) | 4 (50) |
| Plaquetas, n (%) | |||
| <5.000/mm3 | 4 (12,9) | 4 (17,4) | 0 |
| ≥5.000/mm3 | 27 (87,1) | 19 (89,6) | 8 (100) |
| Leucocitos totales | |||
| Media | 9.891 | 10.560 | 7.323 |
| Mediana | 10.100 | 10.700 | 7.045 |
| Intervalo | 4.900-15.700 | 4.900-15.700 | 4.900-11.150 |
| Leucocitos, n (%) | |||
| <6.000/mm3 | 6 (19,4) | 2 (8,7) | 4 (50) |
| ≥6.000/mm3 | 25 (80,6) | 21 (91,3) | 4 (50) |
| Tratamiento, n (%) | |||
| No | 3 (9,7) | 3 (13) | 0 |
| Sí | 28 (90,3) | 20 (87) | 8 (100) |
| Tipo de tratamiento, n (%) | |||
| IGIV sólo | 5 (16,1) | 5 (21,7) | 0 |
| Prednisona sólo | 8 (25,8) | 5 (21,7) | 3 (37,5) |
| IGIV+prednisona | 12 (38,7) | 10 (43,5) | 2 (25) |
| IGIV+prednisona+otros | 3 (9,7) | 0 | 3 (37,5) |
| Esplenectomía | 3 (9,7) | 1 (4,3) | 2 (25) |
IGIV: inmunoglobulina intravenosa.
Variables con significación estadística asociadas a trombocitopenia inmune primaria crónica (más de 12 meses de evolución)
| ≤12 meses (n=23) | >12 meses (n=8) | p | |
|---|---|---|---|
| Edad en años | |||
| Media | 3,70 | 7 | 0,01 |
| <10 años | 22 | 5 | 0,01 |
| ≥10 años | 1 | 3 | |
| Leucocitos totales | |||
| Media | 10.560 | 7.323 | 0,01 |
| <6.000/mm3 | 2 | 4 | 0,01 |
| ≥6.000/mm3 | 21 | 4 | |
| Infección previa | |||
| No | 10 | 8 | 0,005 |
| Sí | 13 | 0 | |
Respecto al tipo de atención recibida por los pacientes con PTI, en 13 casos el diagnóstico fue hecho antes del inicio de la consulta especializada en Hematología, y en los 18 restantes, tras la puesta en marcha de la misma.
En el análisis de regresión logística binaria, empleando como variable dependiente el tipo de PTI (aguda/persistente o crónica), no se encontró ninguna asociación significativa con las variables independientes: sexo, edad, leucocitos y plaquetas al diagnóstico, presencia de sangrado mucoso y tratamiento recibido.
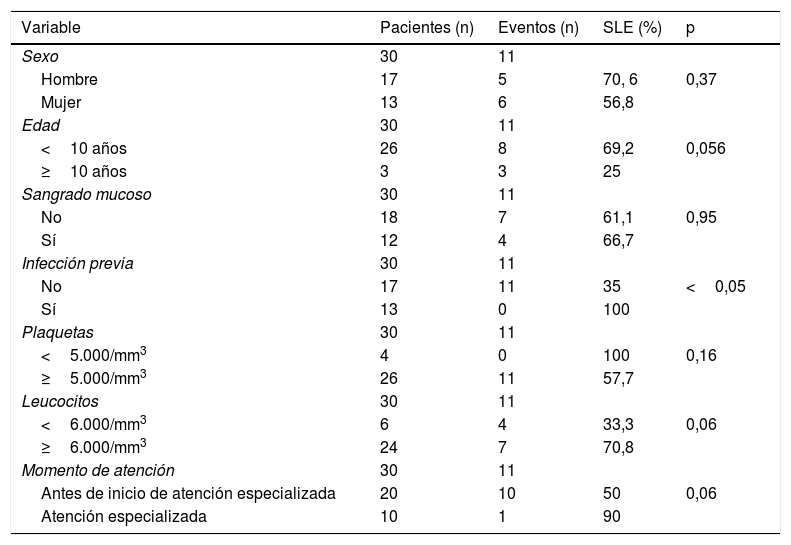
Se comparó la SLE en función de varios factores epidemiológicos, clínicos y terapéuticos, encontrándose que fue menor, aunque sin alcanzar significación estadística, en las mujeres (p=0,37), en los mayores de 10 años (p=0,056), sin signos de sangrado mucoso (p=0,95), con cifras de plaquetas superiores a 5.000/mm3 (p=0,16), con leucopenia <6.000/mm3 (p=0,06), y tratados antes del inicio de la atención especializada en Hematología (p=0,06). La SLE fue significativamente más baja (p=0,001) en los pacientes sin antecedentes de proceso infeccioso previo (tabla 4 y figs. 1-4).
Supervivencia libre de eventos (SLE) en función de factores clínicos, analíticos y según el momento de la atención recibida
| Variable | Pacientes (n) | Eventos (n) | SLE (%) | p |
|---|---|---|---|---|
| Sexo | 30 | 11 | ||
| Hombre | 17 | 5 | 70, 6 | 0,37 |
| Mujer | 13 | 6 | 56,8 | |
| Edad | 30 | 11 | ||
| <10 años | 26 | 8 | 69,2 | 0,056 |
| ≥10 años | 3 | 3 | 25 | |
| Sangrado mucoso | 30 | 11 | ||
| No | 18 | 7 | 61,1 | 0,95 |
| Sí | 12 | 4 | 66,7 | |
| Infección previa | 30 | 11 | ||
| No | 17 | 11 | 35 | <0,05 |
| Sí | 13 | 0 | 100 | |
| Plaquetas | 30 | 11 | ||
| <5.000/mm3 | 4 | 0 | 100 | 0,16 |
| ≥5.000/mm3 | 26 | 11 | 57,7 | |
| Leucocitos | 30 | 11 | ||
| <6.000/mm3 | 6 | 4 | 33,3 | 0,06 |
| ≥6.000/mm3 | 24 | 7 | 70,8 | |
| Momento de atención | 30 | 11 | ||
| Antes de inicio de atención especializada | 20 | 10 | 50 | 0,06 |
| Atención especializada | 10 | 1 | 90 |
El valor de p se ha obtenido mediante el test log-rank.
 en función del antecedente de infección previa (p=0,001).")
 en función de la edad (p=0,056).")
 en función de la cifra de leucocitos (p=0,06).")
 en función del momento de la atención (p=0,06).")
De los 31 pacientes estudiados, 19 (61,3%) fueron dados de alta de la consulta, tras al menos un año de seguimiento después de la resolución de la trombocitopenia. Doce pacientes continuaron seguimiento en la consulta de Hematología Pediátrica, bien por la cronicidad de su cuadro, o bien porque a fecha de fin del estudio no se había cumplido un año de seguimiento.
DiscusiónLos datos epidemiológicos obtenidos en este estudio coinciden con la literatura y los recogidos en diferentes registros en cuanto a la distribución por edad y sexo de los casos de PTI, siendo el grupo etario más frecuente los varones de entre 2 y 4 años3,5. La tasa de incidencia obtenida de 2,6 casos por 100.000 pacientes pediátricos por año se encuentra por debajo de los 5 casos por 100.000 por año que se estima en la mayor parte de las publicaciones3,5. Esta baja estimación de los casos puede atribuirse a varios factores: el primero, que no todos los casos de PTI en Aragón se controlaron en el centro donde se realizó el estudio. Además, en la presente serie, la mayoría de los pacientes tenían una trombocitopenia grave (media de 11.000/mm3, cénit de 47.000/mm3), no habiéndose registrado los casos con trombocitopenia leve, que también cumplirían los criterios diagnósticos de PTI. Si se hubiesen incluido estos pacientes con un cuadro menos sintomático, probablemente la tasa de incidencia se acercaría más a la encontrada en otras publicaciones3,5.
La frecuencia de complicaciones hemorrágicas relevantes es similar a las publicadas (3%), y la de sangrados graves es aún menor que la descrita en la literatura3,6,9. No se registró ningún caso de sangrado potencialmente mortal durante el seguimiento. Dos niños presentaron anemia en relación con los sangrados, precisando solamente uno la administración de hemoderivados. Estos datos avalan la afirmación de que la PTI en la infancia es una enfermedad benigna, y que rara vez asocia morbimortalidad.
La PTI es frecuentemente un trastorno autolimitado que dura pocas semanas o meses, aunque en aproximadamente el 25-30% se convierte en crónica3,13,14. En esta serie, la mayoría de los pacientes alcanzaron una cifra de plaquetas normales en menos de 6 meses y más de dos tercios de la muestra presentaron un cuadro de menos de 12 meses de evolución.
No se encontraron diferencias entre las terapias recibidas. Lo más reseñable fue que los 3 pacientes que no recibieron ningún tratamiento evolucionaron a la curación espontánea en menos de 6 meses. La interpretación de estos resultados está supeditada al pequeño tamaño muestral, pero parece reafirmar que la evolución de este trastorno hacia la curación o cronicidad no depende del tratamiento administrado3,10.
Discernir qué factores pueden influir en la evolución de este trastorno se ha convertido en un tema principal de estudio en los pacientes con PTI9-14, ya que cuando el médico se enfrenta a un diagnóstico nuevo, no puede determinar si el niño va a presentar una enfermedad autolimitada o va a evolucionar a un trastorno crónico, con las implicaciones que esta información tiene de cara al manejo del paciente y su entorno.
En esta serie los pacientes diagnosticados de PTI crónica eran más frecuentemente mujeres, con una edad media más elevada, sin trombocitopenia grave, con leucopenia y sin antecedente de infección previa en la anamnesis al diagnóstico. Se encontró una asociación estadísticamente significativa entre la PTI crónica y la edad más elevada, la presencia de leucopenia y la ausencia de antecedente de infección.
El estudio de regresión no aportó resultados con significación estadística, probablemente por el reducido tamaño muestral, pero en el estudio de supervivencia se confirmó la ausencia de un antecedente infeccioso como un factor con significación estadística para la recaída. La explicación podría ser que, al existir un claro desencadenante infeccioso del proceso autoinmune, el comportamiento sería el clásico y esperado, con una evolución benigna y autolimitada. La ausencia de este desencadenante infeccioso podría ser más sugestivo de que el proceso fisiopatológico que desencadena el fenómeno de disregulación inmune es más complejo y multifactorial, con una tendencia a la cronificación del cuadro. Otros factores que se asociaron a una supervivencia menor, sin llegar a alcanzar la significación, aunque con cifras cercanas a ella, fueron la mayor edad, la presencia de leucopenia y la atención no especializada en Hematología.
ConclusionesLos datos epidemiológicos y de evolución de nuestra muestra coinciden con los de otros registros: la mayoría de los pacientes son varones, de edad preescolar, con antecedente de infección previa, con signos de sangrado mayoritariamente cutáneos, una trombocitopenia grave al diagnóstico y una evolución benigna y menor de 12 meses.
Los pacientes que sean mujeres, de edad escolar o mayor, sin un claro antecedente infeccioso, sin signos de sangrado mucoso, con una trombocitopenia no muy grave y/o leucopenia iniciales, parecen presentar un riesgo mayor de tener una evolución tórpida y recurrente, con tendencia a la cronificación. Dada la baja prevalencia de esta enfermedad sería recomendable realizar un estudio multicéntrico prospectivo con mayor tamaño muestral que permitiese corroborar estos datos con mayor precisión y significación.
En este estudio la evolución de los pacientes fue más favorable a partir del inicio de la atención especializada en Hematología. Con los datos obtenidos, teniendo en cuenta las limitaciones del estudio, parece adecuado recomendar que las enfermedades poco frecuentes, como la PTI, sean manejadas en unidades especializadas, con el objetivo de disminuir la variabilidad en la práctica clínica y ofrecer el tratamiento más adecuado, basado en la más actual y contrastada evidencia científica.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
