





Introducción
Las mitocondrias son organelas citoplasmáticas cuya función principal es llevar a cabo el metabolismo oxidativo, con la consiguiente producción de energía en forma de trifosfato de adenosina (ATP)1. Aunque las mitocondrias poseen un sistema genético propio, no son autónomas, ya que tanto para su formación como para la expresión de su genoma, dependen de un gran número de proteínas codificadas en el núcleo, sintetizadas en los ribosomas citoplasmáticos e importadas a la mitocondria. Por lo tanto, su función está bajo el control de los dos sistemas genéticos celulares: el nuclear y el mitocondrial (comunicación intergenómica)2.
El síndrome de Leigh3 se origina por un trastorno nuclear o mitocondrial genéticamente determinado de aparición esporádica o con herencia variable (autosómica recesiva, ligada al cromosoma X, o herencia materna), que produce un déficit del complejo piruvato-deshidrogenasa y/o déficit de los complejos I-IV de la cadena respiratoria mitocondrial (tabla 1). Afecta de forma heterogénea a diversos órganos y se caracteriza por crisis convulsivas, retraso psicomotor, atrofia óptica, hipotonía, debilidad, letargia, vómitos, movimientos anormales (ataxia, temblor), signos piramidales, irritabilidad, nistagmo, oftalmoplejía externa, pérdida de visión así como anormalidades respiratorias4,5. Presenta mal pronóstico y carece de tratamiento eficaz, aunque se ha utilizado tiamina, coenzima Q10, bicarbonato sódico, dicloroacetato, perfusión de THAM intravenosa y alopurinol, sin éxito apreciable1,6,7.
Observación clínica
Lactante varón de un mes que fue llevado a urgencias por presentar, en ausencia de fiebre, varios episodios breves de hipertonía, tremulación espontánea, revulsión ocular y nistagmo vertical, asociados a hipotonía intercrítica, irritabilidad ocasional y succión débil.
Entre los antecedentes familiares, destacaba padre con espondilitis anquilopoyética. Hermana de 6 años asintomática.
Se registraron los siguientes antecedentes personales: embarazo con amniocentesis normal; parto eutócico a las 40 semanas de edad gestacional; Apgar 9/10; peso de recién nacido: 3.660 kg; no reanimación; fractura de clavícula izquierda. El niño fue ingresado al quinto día de vida por hiperbilirrubinemia indirecta patológica grave con bilirrubina total: 28,9 mg/dl; bilirrubina directa: 0,6 mg/dl, grupo materno O+, grupo recién nacido A+, Coombs directo negativo, estudio inmunohematológico negativo, proteína C reactiva negativa, cultivo de orina negativo, hemograma normal y potenciales evocados auditivos normales. Precisó fototerapia intensiva durante 4 días con evolución favorable, sin que se realizara exanguinotransfusión por disminución rápida de los valores de bilirrubina en las primeras 8 horas a cifras inferiores a 20 mg/dl.
Exploración física al ingreso: temperatura, 36,5 °C; peso, 4,680 kg (P50); talla, 53,5 cm (P50); perímetro cefálico, 39 cm (P90). Buen estado general. Normoconfigurado. Exploración neurológica: fontanela normotensa, pupilas isocóricas y normorreactivas, hiperreflexia osteotendinosa, hipotonía axial, tremulación ocasional de mentón y extremidades, motilidad ocular conservada, irritabilidad ante estímulos, aunque contactaba adecuadamente con el medio. El resto de la exploración por aparatos resultó normal.
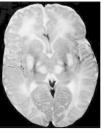
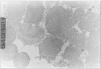
Exploraciones complementarias: hemograma normal; velocidad de sedimentación globular (VSG), 14 mm; bioquímica con normalidad de glucosa, calcio e iones; bilirrubina total, 5,2 mg/dl; bilirrubina directa, 1,1 mg/dl; GOT, 100 U/l; fosfatasa alcalina, 993 U/l, lactato deshidrogenasa (LDH), 708 U/l. Equilibrio acidobásico: pH, 7,33; presión parcial de dióxido de carbono (pCO2), 57; y de oxígeno (pO2), 50; bicarbonato, 30; exceso de bases, 4. Citoquímica y cultivo de líquido cefalorraquídeo (LCR): negativos. Enolasa neuroespecífica en LCR: 32 μg/ml (aumentada). Proteína básica de mielina en LCR: 0,3 ng/ml (0-1,5 ng/ml). Perfil de aminoácidos en sangre y orina: normales. Ácido láctico, 26,1 mg/dl (3-15 mg/dl), y ácido pirúvico, 1,6 mg/dl (0,3-1 mg/dl), aumentados en sangre. Cociente lactato/piruvato, 16. Ácido acetoacetato y 3-hidroxibutirato, aumentados. Ecografía cerebral, normal. Fondo de ojo, normal. Electroencefalograma: actividad de fondo lentificada con puntas aisladas parietotemporales izquierdas. Resonancia magnética (RM) cerebral: alteraciones de intensidad de señal (aumento en T2) bilateral y simétrica en núcleos lenticulares, tálamos, núcleos rojos mesencefálicos y protuberancia así como vermis inferior, con ausencia de mielinización en sustancia blanca de circunvoluciones pre y poscentral (fig. 1). Biopsia del músculo estriado: fibras tipo 1 con disminución anular subsarcolémica de la actividad oxidativa. Al microscopio electrónico: ausencia de fibras rojas rasgadas y presencia de fibras musculares con mitocondrias de tamaño aumentado compatible con probable anomalía mitocondrial (fig. 2). Estudio de función mitocondrial en homogenado muscular: déficit de los complejos I, III y IV de la cadena respiratoria (tabla 2).
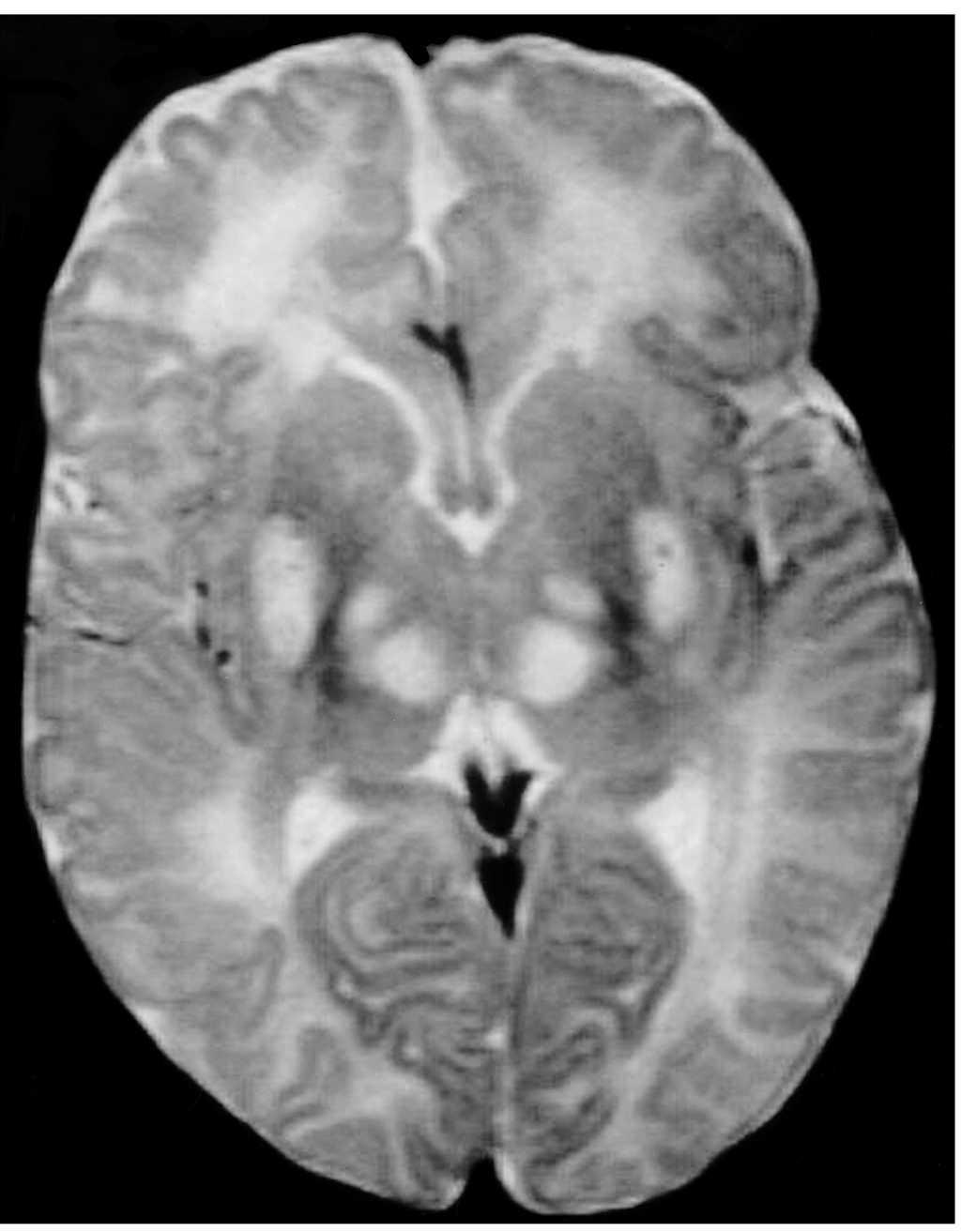
Figura 1.RM cerebral. Corte axial: aumento de intensidad en T2 bilateral y simétrica de la sustancia blanca periventricular y núcleos de la base (tálamos y lenticular).
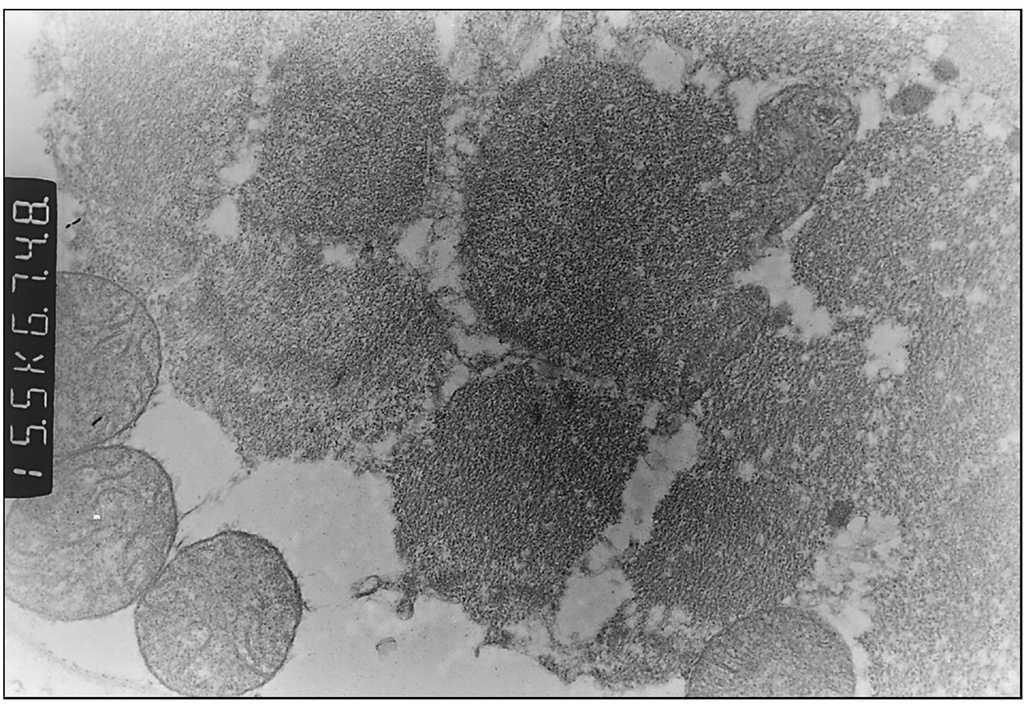
Figura 2. Biopsia muscular: aspecto con microscopia electrónica. Megamitocondrias (0,9μ) de tamaño algo inferior a las miofibrillas (x31.000).

Evolución y tratamiento: desde el ingreso presentó crisis progresivamente más frecuentes con tendencia al coma pese a la administración de diazepam y fenobarbital intravenosos, por lo que fue trasladado a la unidad de cuidados intensivos pediátricos del Hospital Infantil La Paz, precisando sedación con perfusión continua de midazolam, así como soporte respiratorio, hemodinámico y nutricional, presentando como procesos intercurrentes anemia, infección urinaria y gastroenteritis aguda. Desarrolló deterioro neurológico rápidamente progresivo que condujo al coma irreversible y muerte a la edad de 2 meses.
Discusión
La cadena respiratoria se encuentra situada en la membrana interna mitocondrial, y está integrada por una serie de complejos enzimáticos (I al V) que contienen aproximadamente 70 polipéptidos, y dos moléculas que actúan a modo de nexos de unión (coenzima Q y citocromo C).
El ADN mitocondrial (ADNmt) es una molécula circular de doble hebra que procede exclusivamente del óvulo1. Contiene solamente 37 genes, 13 de los cuales codifican polipéptidos de la cadena respiratoria mitocondrial (el complejo II y los demás polipéptidos de los otros cuatro complejos están bajo el control del genoma nuclear). Los otros 24 genes son necesarios para la traducción de proteínas en los ribosomas mitocondriales: 22 codifican ARN de transferencia (ARNt) y 2 ARN ribosomal (ARNr). Cada mitocondria contiene de 2 a 10 copias de ADNmt. Las mutaciones en el ADNmt pueden afectar a todos los genomas o a parte de ellos, y puede coexistir en la misma célula o tejido ADNmt normal y mutado (heteroplasmia). La expresión fenotípica de una mutación patogénica del ADNmt no sigue las reglas de la herencia mendeliana y depende del efecto umbral o proporción mínima de ADNmt mutado necesaria para alterar el metabolismo oxidativo, así como de las demandas energéticas en cada momento de los distintos órganos o tejidos1,2.
Las alteraciones del ADNmt incluyen las deleciones únicas (habitualmente esporádicas), las duplicaciones/deleciones simples y las mutaciones puntuales (ambas de herencia materna). En este tipo de herencia, una madre que porte una mutación en su ADNmt, puede transmitirla a sus hijos e hijas, pero sólo sus hijas la transmitirán a la descendencia. Las alteraciones dependientes del ADNn tienen herencia mendeliana e incluyen: alteraciones de los genes nucleares que codifican proteínas mitocondriales, alteraciones en la importación de proteínas mitocondriales y alteraciones en la comunicación intergenómica (deleciones múltiples del ADNmt y depleción del ADNmt).
En las encefalomiopatías mitocondriales, un idéntico fenotipo clínico puede relacionarse con anomalías bioquímicas o moleculares diferentes y viceversa2. Así, las manifestaciones clínicas del síndrome de Leigh no siempre indican disfunción de la cadena respiratoria y su sustrato genético puede ser variable (tabla 3).
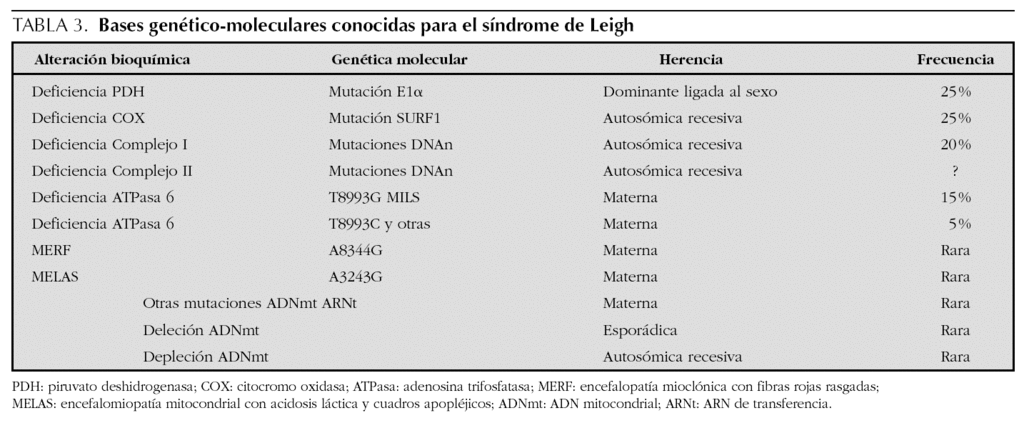
La sintomatología clínica en nuestro paciente se puede corresponder con la forma congénita del déficit del complejo I (enfermedad multisistémica precoz y fatal caracterizada por acidosis láctica, convulsiones, hipotonía, debilidad muscular, cardiopatía y muerte en el período neonatal por parada cardiorrespiratoria), con la encefalomiopatía infantil fatal por déficit del complejo III (acidosis láctica, hipotonía, convulsiones, coma y muerte en los primeros meses) (tabla 1), y con el síndrome de Leigh con herencia materna (MILS), que tiene su inicio en los primeros meses de vida, un curso evolutivo rápido conduciendo a la muerte en el primer año3-5,8, y cuya base molecular consiste en la mutación puntual T8993G del gen que codifica la subunidad 6 de la ATPasa, con una proporción de ADNmt mutado que supera al 90 % de los genomas mitocondriales1. En nuestro caso, el diagnóstico de la enfermedad fue posible gracias a la asociación de un cuadro clínico compatible, con unos valores de lactato y piruvato séricos elevados9,10, así como la presencia en la resonancia magnética (RM) cerebral de incrementos de intensidad de señal en T2 localizados simétricamente en áreas típicamente afectadas11-13. Los hallazgos neuropatológicos no pudieron ser confirmados ante la negativa de los padres para autorizar la necropsia. Pensamos que la grave hiperbilirrubinemia que presentó en el período neonatal precoz pudiera ser la manifestación de una disfunción hepática producida como consecuencia del trastorno mitocondrial a ese nivel6. Por otra parte, tal como se describe en la literatura especializada, las crisis convulsivas con signos electroencefalográficos de focalidad fueron la forma de presentación inicial de la enfermedad14. Los hallazgos histopatológicos de la biopsia muscular orientaron hacia una anomalía mitocondrial15. Finalmente, el estudio bioquímico en músculo esquelético de los distintos complejos de la cadena respiratoria mitocondrial, condujo a la detección del triple déficit12,16.
Por otra parte, los defectos únicos de los complejos de la cadena respiratoria son más característicos de las alteraciones del ADNn, mientras que los defectos combinados son habituales en las mutaciones del ADNmt1. Actualmente se está realizando en nuestro paciente el estudio genético de las diferentes mutaciones del ADNmt. La deficiencia de tres complejos codificados parcialmente por el ADNmt apoya la posibilidad de que su base genética radique en una mutación puntual en alguno de los 22 genes que codifican ARNt, en una deleción simple del ADNmt o bien en una alteración en la comunicación intergenómica (deleciones múltiples y/o depleción del ADNmt)13,17-21. Desafortunadamente, el estudio genético-molecular es poco útil para la detección precoz intraútero y el consejo genético familiar, dado que la genética mitocondrial no sigue un patrón mendeliano6,22. Por último, queremos señalar que debido a la precocidad y rapidez evolutiva con que se presentó y cursó la enfermedad no fue posible instaurar una terapia de prueba que muy probablemente hubiera sido ineficaz6,7.
Agradecimientos
Los autores agradecen la inestimable colaboración del Dr. J. Arenas y la Dra. Y. Campos, del Centro de Investigación del Hospital 12 de Octubre de Madrid, para el estudio del metabolismo mitocondrial del paciente presentado, así como del Dr. Gutiérrez, del Servicio de Anatomía Patológica (Neuropatología) del Hospital La Paz de Madrid, por la cesión de las imágenes de microscopia electrónica.
