



Introducción
El síndrome de Aicardi-Goutières es una enfermedad neurodegenerativa de herencia autosómica recesiva1, que se caracteriza por presentar encefalopatía grave y progresiva de inicio precoz, microcefalia evolutiva, desarrollo de tetraplejía espástica y afectación del sistema extrapiramidal, calcificaciones intracraneales predominantemente a nivel de ganglios basales, afectación de la sustancia blanca, linfocitosis y grados variables de hiperproteinorraquia, así como elevación del interferón a (IFN-α) en sangre y de manera más importante en líquido cefalorraquídeo (LCR)2,3.
El objetivo es presentar dos nuevas observaciones, diagnosticadas en función de un curso clínico compatible, hallazgos neurorradiológicos e inmunológicos característicos, en ausencia de evidencias de infección congénita causal.
Casos clínicos
Paciente 1
Niño de 3 meses de edad, que consultó por presentar, desde el nacimiento, vómitos frecuentes, rechazo de alimentación e irritabilidad. Segundo hijo de padres consanguíneos en tercer grado. Embarazo a término, parto vaginal espontáneo, Apgar 9-10. Datos somatométricos al nacimiento: peso, 2.630 g; talla, 45,5 cm, y perímetro craneal (PC), 31 cm (< 2 DE). En la exploración física se observaba microsomía armónica, signos de desnutrición, irritabilidad, ausencia de contacto ocular, nistagmo horizontal y tono cefálico deficiente. Datos somatométricos: peso, 3.670 g (< P10); talla, 52 cm (< P10) y PC 36,5 cm (P < 2 DE).
Entre las exploraciones complementarias realizadas destacaban: gasometría, inmunoglobulinas plasmáticas, perfil tiroideo, ácido pirúvico plasmático, amoniemia, catecolaminas y test de sulfitos en orina, determinación de aminoácidos en sangre, orina y LCR, ácidos orgánicos en orina y LCR, perfil de acilcarnitinas, cuerpos cetónicos plasmáticos, fondo de ojo, serie ósea, ecografía abdominal, electromiografía y estudio neurográfico, normales. Se observó ligero aumento de transaminasas e hiperlactacidemia sostenida. Los estudios neurofisiológicos demostraron importante afectación tanto de la vía auditiva como de la visual, mediante la realización de potenciales evocados auditivos tronculares (PEAT) y visuales (PEV), así como un trazado desorganizado y elementos agudos difusos en el electroencefalograma (EEG). La neuroimagen puso de manifiesto calcificaciones en los ganglios basales, afectación grave de la sustancia blanca e imágenes de lisencefalia. Se demostró la negatividad de serologías TORCH (toxoplasmosis, rubéola, citomegalovirus, herpes) en sangre y LCR, destacando la presencia en dos determinaciones de LCR de hiperlactorraquia, elevación del ácido pirúvico, ligero aumento de celularidad de predominio linfocítico en la primera y moderada hiperproteinorraquia en ambas muestras (1,1 y 1,3 g/l, respectivamente).
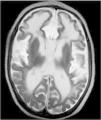
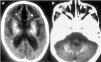
Evolutivamente se observó escaso incremento del perímetro craneal, desconexión medioambiental, desarrollo de tetraplejía espástica, control cefálico deficiente e hipertonía generalizada. Se realizó nueva neuroimagen (tomografía computarizada [TC] y resonancia magnética [RM]), donde se observaron extensas calcificaciones bilaterales cerebrales supratentoriales e infratentoriales, afectándose ambos hemisferios cerebelosos, núcleos basales y zona periventricular, ausencia de mielinización, lisencefalia frontal e intensa atrofia corticosubcortical (figs. 1 y 2). Se realizó biopsia muscular del vasto externo, cuyo estudio anatomopatológico y enzimático de la cadena respiratoria mitocondrial fue normal. Se demostró elevación del IFN-α en plasma y más marcado en LCR (12 y 200 U/ml, respectivamente, valores normales < 2 U/ml). El paciente desarrolló epilepsia y presentó nula adquisición psicomotora, y murió a los 27 meses de edad.
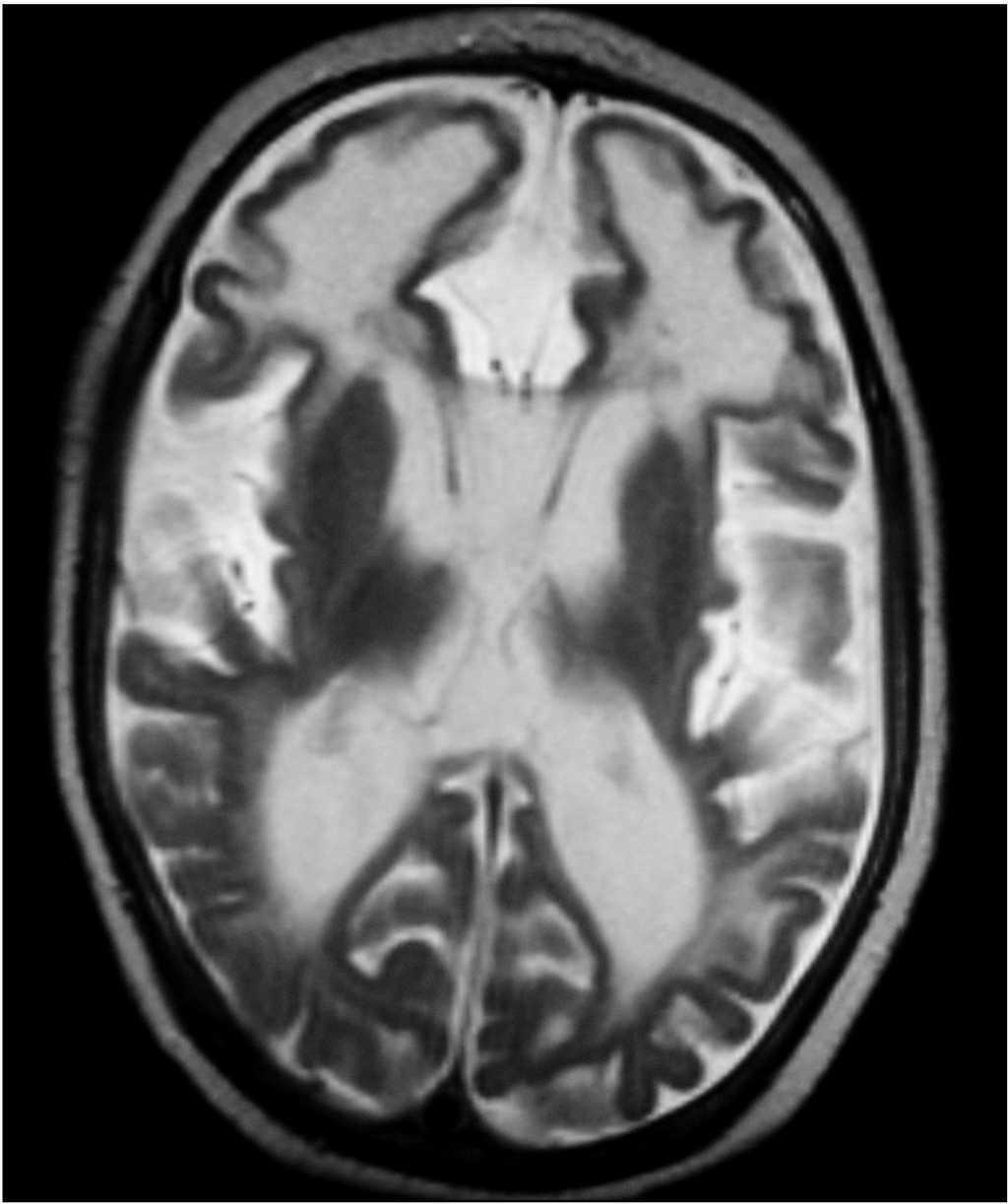
Figura 1.RM cerebral. Imagen axial (potenciación T2), con lisencefalia frontal, intensa atrofia corticosubcortical y alteración de la mielinización.
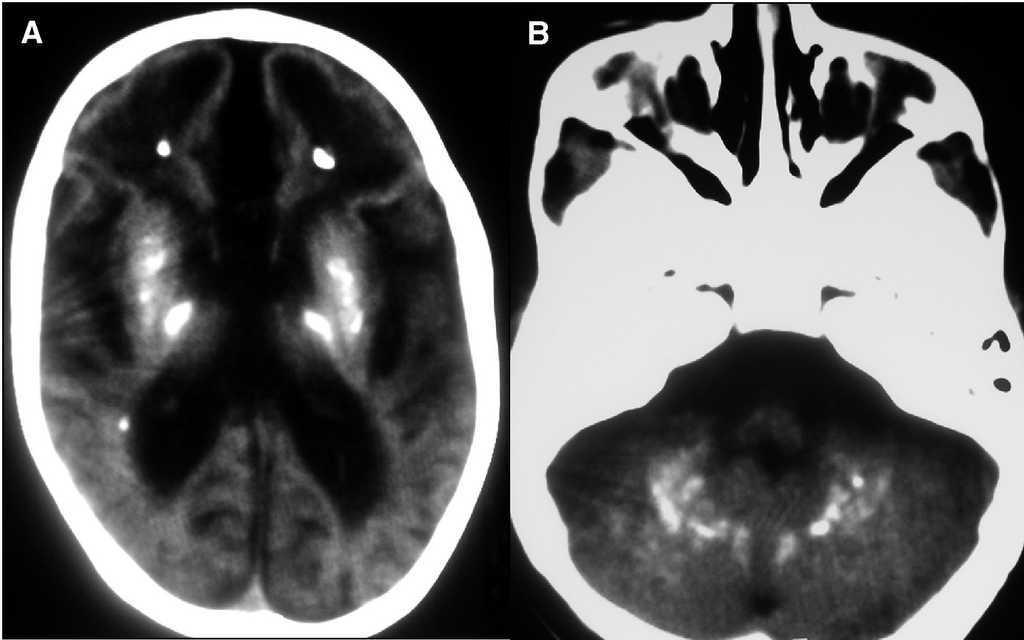
Figura 2.A)TC cerebral. Extensas calcificaciones bilaterales cerebrales que afectan núcleos basales y zona periventricular.B)TC cerebral. Calcificaciones en hemisferios cerebelosos.
Paciente 2
Niña de 9 meses de edad, que fue traída a consulta por presentar retraso psicomotor. Era la primera hija de padres no consanguíneos. Embarazo a término; parto vaginal espontáneo; Apgar 9-10. Datos somatométricos al nacimiento: peso, 2.430 g (< P10), longitud 45 cm (< P10) y perímetro craneal (PC) de 34 cm (P25). Consta la presencia de dificultades para la alimentación en el período neonatal.
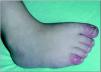
En la exploración física en el momento de la evaluación inicial presentaba un patrón de inversión fisiológica del tono muscular, con control cefálico deficiente, ausencia de sedestación y leve espasticidad isquiotibial y gastrosólea. Seguimiento ocular presente con nistagmo de fijación y estrabismo convergente. Evolutivamente asocia posiciones distónicas en las cuatro extremidades, movimientos coreicos y lesiones de eritema pernio en pies y manos (fig. 3). Somatometría: peso y talla en P3; PC en 2 DE, con estancamiento posterior situándose a los 23 meses en 3,5 DE.
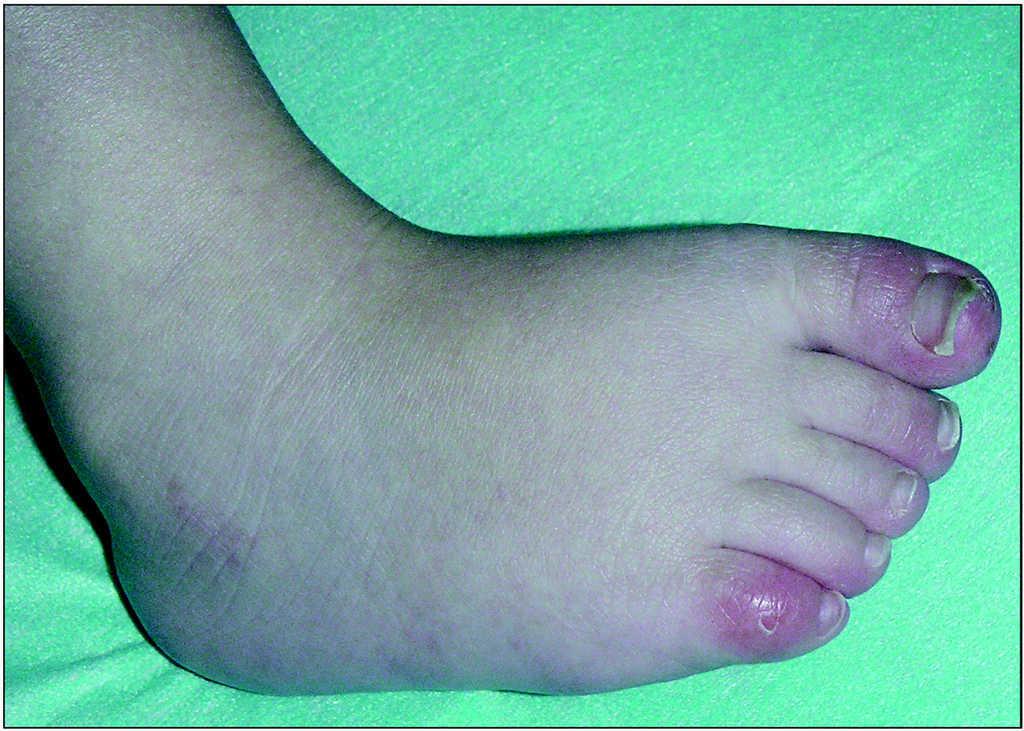
Figura 3. Lesiones de eritema pernio en pies.
Entre las exploraciones complementarias realizadas destacan: transaminasemia, amoniemia, ácido láctico y pirúvico plasmático, aminoácidos en sangre y orina, ácidos orgánicos en orina, estudio citoquímico convencional de LCR, serologías TORCH, cariotipo, PEAT, EEG y fondo de ojo, normales; PEV desestructurados. La RM cerebral realizada a los 11 meses de edad mostraba lesiones quísticas periventriculares, signos de atrofia cortical y aumento de la señal en la sustancia blanca periventricular. La TC craneal realizada a los 24 meses de edad revelaba calcificaciones bilaterales en los núcleos de la base y alguna periventricular, con disminución global del grosor de la sustancia blanca. El IFN-α en LCR y sangre mostró valores elevados (4 y 25 U/ml, respectivamente).
Discusión
El cuadro clínico de los 2 pacientes fue similar al descrito clásicamente por Aicardi y Goutières4, y cumplían todos los criterios diagnósticos del síndrome2,5 (tabla 1), siendo encuadrables en una forma grave6.

El diagnóstico se estableció ante la asociación de los siguientes hechos:
1. Cuadro clínico caracterizado por nula adquisición de hitos del desarrollo psicomotor, desconexión medioambiental, desarrollo de importante microcefalia y presencia de movimientos oculares anormales.
2. Grandes dificultades en la alimentación acompañado de desnutrición.
3. Evolución hacia tetraplejía espástica con disfunción extrapiramidal.
4. Hallazgo en neuroimagen de extensas calcificaciones, ausencia de mielinización y signos de atrofia difusa.
5. Pleocitosis de predominio linfocítico en LCR acompañado de moderada hiperproteinorraquia en la primera observación.
6. Elevación del IFN-α en plasma y en LCR.
7. Cribado negativo de infecciones antenatales.
El síndrome de Aicardi-Goutières es una enfermedad neurodegenerativa, de inicio precoz y curso clínico grave y progresivo, aunque se han descrito recientemente observaciones con afectación más moderada7 y de inicio más tardío8, con amplia variabilidad en la presentación en miembros de una misma familia9. Se transmite con herencia autosómica recesiva1, demostrándose mutaciones a nivel del locus 3p21 en la mitad de los casos10. Desde su primera descripción en 19844 se han notificado cerca de 70 observaciones5, aunque probablemente la prevalencia real esté infraestimada, debido en parte a la confusión existente en sus criterios diagnósticos y al solapamiento con otras entidades.
Se caracteriza por un inicio precoz, antes del primer año de vida y con frecuencia desde el nacimiento2 la mayoría de veces sin antecedentes obstétricos ni neonatales destacables, estando descritos casos con desarrollo psicomotor inicial normal7 de encefalopatía progresiva, precedido por frecuente irritabilidad y dificultades para la alimentación, desarrollo de microcefalia evolutiva, enlentecimiento en el desarrollo psicomotor seguido de regresión neurológica o bien nula adquisición desde su inicio, desarrollo de tetraplejía espástica y afectación del sistema extrapiramidal, frecuente alteración en los movimientos oculares, epilepsia en el 20 % de los casos y fallecimiento en los primeros años de vida5. Como hechos extraneurológicos pueden presentar alteraciones cutáneas en forma de exantema eritematoso descamativo y acrocianosis, principalmente en dedos de manos y pies, aunque también en pabellones auriculares, remedando al eritema pernio11; hepatomegalia, elevación moderada de transaminasemia y trombocitopenia.
La presencia de calcificaciones en los ganglios basales, más frecuentes a nivel del putamen, constituye un criterio diagnóstico mayor, y puede afectar también al resto de corteza y cerebelo. Ocasionalmente este dato puede estar ausente en los estadios iniciales y demostrarse posteriormente. De manera habitual se observa afectación de la sustancia blanca hemisférica11,12 de predominio frontal y periventricular, y atrofia generalizada corticosubcortical, de evolución variable13.
Otro criterio diagnóstico lo constituye el hallazgo de pleocitosis (habitualmente entre 20 y 80 cél./μl) de predominio linfocítico (> 80 %) a nivel de LCR, dato dependiente de la edad, pudiendo estar ausente en los mayores de un año de edad como ocurrió en la segunda observación2; se asocian, aunque en un porcentaje inferior, grados variables de hiperproteinorraquia. El IFN-α es una citoquina inmunomoduladora que interviene en los mecanismos de defensa antivirales y antitumorales. Desde el hallazgo de la presencia de títulos altos de IFN-α en sangre y de manera más importante en LCR en esta entidad3,14, su demostración ha pasado a ser un criterio diagnóstico principal, habitualmente presente al menos en los estadios iniciales de la enfermedad3.
Existen más de 50 entidades con calcificaciones de los ganglios basales11,15. El diagnóstico diferencial se ha de establecer principalmente con las infecciones congénitas TORCH y especialmente con el citomegalovirus, mucho más frecuentes y susceptibles de beneficio con tratamiento, con todo lo que implica de cara al consejo genético. Existe un grupo de entidades, entre las que se encuentran el síndrome seudo-TORCH16 y la encefalitis de los indios Cree de Quebec17,18, que presentan múltiples datos comunes, incluido retraso psicomotor grave, microcefalia, calcificación de los ganglios basales, e incluso presencia de microangiopatía19, elevación del IFN-α18 e idéntica mutación genética18, formando parte probablemente de una misma entidad con expresión variable. Blau et al20 describieron recientemente una variante del síndrome en 3 pacientes que cursaban sin pleocitosis y con concentraciones normales de IFN-α, en los que se demostró la elevación marcada de LCR de pterinas (neopterina y biopterina) con descenso de la cifra de folatos, alteraciones que se revirtieron en uno de los pacientes con la administración oral de ácido folínico.
El mecanismo fisiopatológico principal que subyace en el desarrollo de las calcificaciones del síndrome de Aicardi-Goutières es una vasculitis que afecta al cerebro y a los vasos sistémicos. El IFN-α interviene con toda probabilidad en la patogenia de la vasculitis mediante mecanismos proinflamatorios3,14 y es causa por sí mismo de encefalopatía14; la causa de su elevación podría residir en una disregulación por mutación de su gen codificador, precipitado o no por una infección viral desconocida21.
En conclusión, a pesar de su relativa rareza, debe sospecharse esta entidad ante un paciente con afectación del desarrollo psicomotor, microcefalia, disfunción pirámido-extrapiramidal y presencia de calcificaciones en el sistema nervioso central. El diagnóstico diferencial con otras entidades más prevalentes, en especial las infecciones TORCH, es esencial de cara al consejo genético.
Agradecimientos
Al Prof. P. Lebón del Servicio de Microbiología del Hospital de Saint Vincent de Paul de París, por la determinación del IFN-α en suero y LCR.
