

La hipotonía asociada a dificultades de alimentación durante el periodo neonatal plantea un diagnóstico diferencial amplio, que implica la realización de numerosas exploraciones complementarias para descartar causas tan diversas como sufrimiento fetal agudo, malformaciones cerebrales, enfermedades neuromusculares o metabólicas o síndromes genéticos.
Varón de 21 meses, hijo de padre español y madre china, seguido en la consulta de neuropediatría desde los 3 meses por hipotonía severa con reflejos osteotendinosos conservados y dificultades para alimentarle desde el periodo neonatal. Requirió sonda nasogástrica hasta los 4 meses de vida.
Antecedentes personales: fruto de una segunda gestación. Nació a término por cesárea, en un centro extranjero, por riesgo de pérdida del bienestar fetal tras un periodo de dilatación prolongado de 50h, con líquido teñido de meconio. El Apgar fue 9/9. Fue macrosómico, con longitud y perímetro craneal por debajo de la media. Presentó hipoglucemia severa sintomática, con crisis convulsiva única a las 12h de vida. A las 24h de vida presentó cianosis y mal estado general, y se diagnosticó coartación de aorta; precisó de cirugía, que cursó sin incidencias. Se detectó además una displasia congénita de caderas bilateral, tratada con férula de abducción.
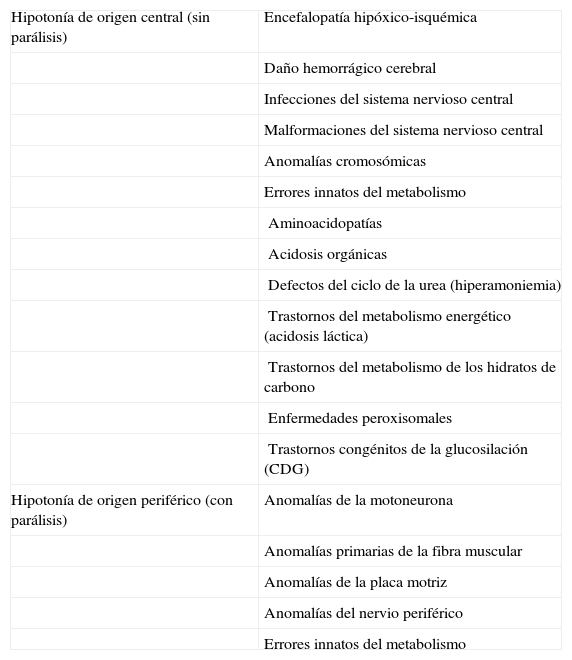
Para descartar las principales enfermedades que cursan con hipotonía durante el periodo neonatal y del lactante (tabla 1), se realizaron diversas pruebas complementarias durante los primeros meses de vida. La resonancia magnética cerebral y de columna a los 3 y a los 11 meses de vida no mostró hallazgos patológicos. Las serologías TORCH, el cariotipo convencional, el estudio FISH para del22q11 y la radiografía de tórax y columna también fueron normales. Los estudios metabólicos no mostraron anomalías e incluyeron función hepática, renal y tiroidea, creatincinasa, amonio, lactato, piruvato, gasometría venosa, aminoácidos y ácidos orgánicos en sangre y orina, ácidos grasos de cadena muy larga, carnitina libre y total, acilcarnitinas y sialotransferrinas. El electroencefalograma, los potenciales evocados auditivos de tronco cerebral, el estudio de velocidad de conducción motriz y sensitiva y el electromiograma fueron normales.
Diagnóstico diferencial de la hipotonía neonatal y del lactante
| Hipotonía de origen central (sin parálisis) | Encefalopatía hipóxico-isquémica |
| Daño hemorrágico cerebral | |
| Infecciones del sistema nervioso central | |
| Malformaciones del sistema nervioso central | |
| Anomalías cromosómicas | |
| Errores innatos del metabolismo | |
| Aminoacidopatías | |
| Acidosis orgánicas | |
| Defectos del ciclo de la urea (hiperamoniemia) | |
| Trastornos del metabolismo energético (acidosis láctica) | |
| Trastornos del metabolismo de los hidratos de carbono | |
| Enfermedades peroxisomales | |
| Trastornos congénitos de la glucosilación (CDG) | |
| Hipotonía de origen periférico (con parálisis) | Anomalías de la motoneurona |
| Anomalías primarias de la fibra muscular | |
| Anomalías de la placa motriz | |
| Anomalías del nervio periférico | |
| Errores innatos del metabolismo |
Presenta un retraso psicomotor global, que mejoró inicialmente con estimulación temprana aunque evoluciona con lentitud, con inicio del sostén cefálico a los 7 meses y sedestación desde los 12 meses, pero aún no ha adquirido la bipedestación a los 21 meses. Ha presentado crisis febriles atípicas y focales secundariamente generalizadas, controladas con ácido valproico. Progresivamente se ha ido constatando en las sucesivas revisiones la presencia de rasgos peculiares en la cara y las manos, propios del síndrome de Kabuki (SK): epicanto bilateral, fisura palpebral larga con eversión de la porción externa de los párpados inferiores, cejas ensanchadas en su porción externa, nariz corta con puente nasal deprimido, labio superior en trapecio con comisuras bucales alargadas hacia abajo, orejas grandes y displásicas con fístula preauricular, paladar ojival, mamilas pequeñas separadas, falanges de manos cortas y abultamiento de pulpejos de todos los dedos de las manos y los pies (fig. 1).

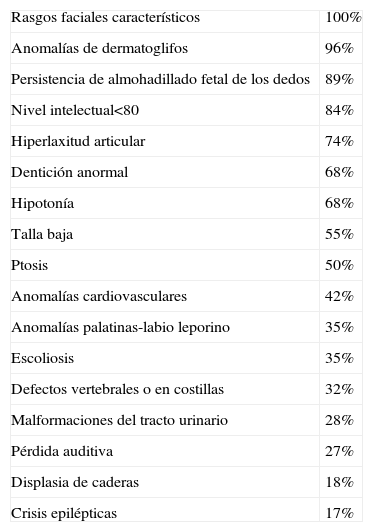
El SK tiene una prevalencia, probablemente subestimada, entre 1/32.000 recién nacidos japoneses1 y 1/86.000 nacidos en Oceanía2. Afecta casi por igual a ambos sexos, y parecen ser en su mayoría casos esporádicos, aunque se han descrito casos familiares con una herencia autosómica dominante3. El diagnóstico por ahora es clínico, basado en los rasgos faciales y corporales específicos definidos inicialmente por Niikawa et al4 y Kuroki et al5, identificables independientemente del origen étnico y que debemos conocer para evitar exploraciones innecesarias. No obstante, es una entidad muy heterogénea en su presentación, con múltiples malformaciones y alteraciones sistémicas descritas en diversas series de casos6,7 (tabla 2). Durante la época neonatal el diagnóstico es difícil, ya que los rasgos faciales más típicos no son tan obvios. Se pueden distinguir en algunos casos las hendiduras palpebrales largas, las orejas grandes mal formadas, la nariz corta con puente deprimido o la persistencia fetal del almohadillado de los dedos, pero no la eversión del párpado inferior lateral, tan característica del SK8. Un 8% puede presentar además hipoglucemia neonatal por hiperinsulinismo9.
Características del síndrome de Kabuki, según Matsumoto et al6, 2003
| Rasgos faciales característicos | 100% |
| Anomalías de dermatoglifos | 96% |
| Persistencia de almohadillado fetal de los dedos | 89% |
| Nivel intelectual<80 | 84% |
| Hiperlaxitud articular | 74% |
| Dentición anormal | 68% |
| Hipotonía | 68% |
| Talla baja | 55% |
| Ptosis | 50% |
| Anomalías cardiovasculares | 42% |
| Anomalías palatinas-labio leporino | 35% |
| Escoliosis | 35% |
| Defectos vertebrales o en costillas | 32% |
| Malformaciones del tracto urinario | 28% |
| Pérdida auditiva | 27% |
| Displasia de caderas | 18% |
| Crisis epilépticas | 17% |
La afección neurológica principal es el retraso mental, casi de forma invariable. La hipotonía durante el periodo neonatal está presente en un 30-100% de los casos descritos, según las series2,3,6–8, con variable grado de afección, que mejora significativamente con la estimulación temprana, con un desarrollo de sedestación hacia los 11 meses de media y marcha autónoma entre los 15 y los 30 meses10. También es importante destacar que los estudios electrofisiológicos y las biopsias musculares realizadas en otros casos publicados no han aportado datos relevantes3. Las dificultades de alimentación son frecuentes durante el primer año y, aunque transitorias, hasta un 29% de los pacientes pueden precisar sonda nasogástrica o incluso gastrostomía2. La causa de esta dificultad parece estar en un componente de hipotonía oral, además de una descoordinación orofaríngea, aunque algunos pacientes presentan alteraciones palatinas añadidas3,9.
Con este caso destacamos que debemos tener en cuenta el diagnóstico de SK en un neonato con hipotonía y dificultades de alimentación, sobre todo si asocia cardiopatía congénita u otras malformaciones. Los rasgos faciales característicos no serán tan obvios en el recién nacido, pero irán acentuándose a lo largo del primer año de vida, y la hipotonía y las dificultades para alimentarse mejorarán progresivamente.
