

La deleción del brazo corto del cromosoma 18 —del(18p)— fue descrita por primera vez por Grouchy, genético francés, en 1963. Desde entonces se han descrito más de 100 casos, y se lo considera uno de los síndromes de deleción cromosómica más frecuentes1. En la mayoría de los casos (85%) se trata de deleciones de novo, que aparecen en una fase precoz del desarrollo embrionario, y es rara la transmisión familiar de del(18p)2.
Se describe el caso clínico de un varón caucásico de 32 meses de edad, orientado para la consulta de pediatría por baja estatura.
Primer hijo de padres jóvenes, no cosanguíneos. La gestación fue vigilada y el parto fue eutócico a las 37 semanas de gestación, hospitalario, sin necesidad de reanimación. Al nacimiento presentaba valores antropométricos compatibles con retraso de crecimiento intrauterino simétrico. La evolución ponderal se situó en P5-P10 hasta los 12 meses, y después se verificó evolución para percentiles por debajo del P5. La estatura y el perímetro cefálico fueron, desde el nacimiento, inferiores al P5.
Presentaba un desarrollo psicomotor adecuado, excepto retraso del inicio del habla.
En el examen físico presentaba peso, estatura y perímetro cefálico inferiores al P5 para la edad y el sexo; microcefalia y braquicefalia; piel facial seca y eccematosa; retrognatia; epicanto bilateral y ligera ptosis palpebral; rarefacción del tercio externo de las cejas; paladar ojival y protrusión de la lengua; orejas prominentes, normalmente implantadas e implantación baja del cabello. La exploración física de las extremidades mostraba clinodactilia de la última falange del quinto dedo de ambas manos, el cuarto dedo supraaducto con clinodactilia de ambos pies, sindactilia ligera del segundo y el tercer dedo de los pies y displasia ungueal del quinto dedo.
En la investigación clínica de este niño, se realizaron los siguientes exámenes complementarios de diagnóstico: hemograma y bioquímica sérica, análisis sumario de orina, estudio inmunológico, función tiroidea, dosificación de hormona de crecimiento, factor de crecimiento similar a la insulina 1 y estudio metabólico, que se revelaron normales. El rastreo inmunológico de la enfermedad celíaca y el examen parasitológico en las heces fueron negativos. La telerradiografía de la mano y la muñeca derechas documentó edad ósea compatible con edad cronológica y la resonancia magnética nuclear cerebral fue normal.
Los estudios cromosómicos de citogenética clásica revelaron una deleción de parte del brazo corto de uno de los cromosomas 18 —46,XY,del(18)(p11.2) mat—, compatible con un diagnóstico de síndrome de 18p- (fig. 1A).
 del paciente (A) y de la madre (B), que muestra el par de cromosomas 18. Se indica el punto de quiebra 18p11.21.")
Se realizaron los estudios cromosómicos de los padres. El padre reveló un cariotipo de constitución cromosómica normal (46,XY) y presentaba 160cm de estatura, con examen físico normal; no había realizado la escolaridad normal.
El cariotipo de la madre evidenció una deleción de parte del brazo corto del cromosoma 18 (fig. 1B), aparentemente igual a la del hijo. A través de la técnica de citogenética molecular (FISH) efectuada en la madre, se excluyó que hubiera material delectivo del brazo corto del cromosoma 18 anormal en cualquier otro cromosoma.
En la observación clínica, la madre presentaba baja estatura (< P5), perímetro craneal en el P10 y peso normal. Presentaba también señales dismórficas idénticas a las del hijo, en concreto braquicefalia, facies inexpresiva, con piel seca y descamativa, oreja izquierda prominente, paladar normal, con acabalgamiento de los dientes incisivos, retrognatia, esbozo de epicanto, ligera ptosis palpebral y rarefacción del tercio externo de las cejas. En el examen de las extremidades, tenía clinodactilia de la última falange del quinto dedo de ambas manos, sindactilia ligera del segundo y el tercer dedo de los pies y displasia ungueal del quinto dedo.
La evaluación neuropsicológica evidenció un cociente intelectual general de 56, con un cociente intelectual de realización de 53 y un cociente intelectual verbal de 64.
Se realizó el estudio del cariotipo a la abuela del niño, que presentaba antecedentes de dos abortos espontáneos en el primer trimestre de gestación, cuyo resultado fue normal (46,XX).
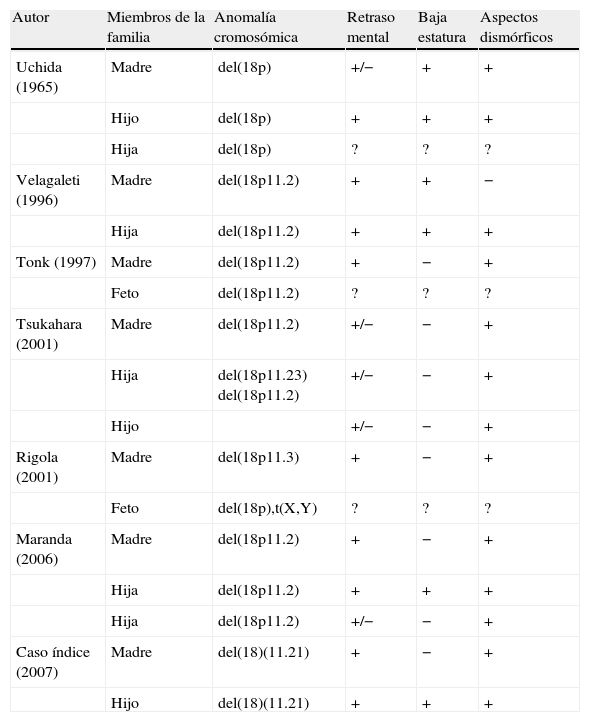
Apenas 6 casos de transmisión familiar están descritos en la literatura: Uchida3 (1965), Velagateli4 (1996), Tonk5 (1997), Tsukahara6 (2001), Rigola7 (2001) y Maranda2 (2006).
Los individuos afectados de la deleción 18p tienen gran variabilidad fenotípica. Cerca del 20% de los recién nacidos no sobreviven debido a malformaciones cerebrales graves. Las manifestaciones en los recién nacidos son sutiles, más evidentes hacia los 3 años de edad.
Característicamente, los individuos afectados de la deleción 18p tienen retraso mental, baja estatura, dismorfias craneofaciales, orejas prominentes, ptosis palpebral, múltiples caries dentarias, cuello corto y alteraciones esqueléticas8. La distonía es frecuente y se manifiesta entre los 12 y los 17 años.
Este caso presenta manifestaciones fenotípicas muy características y descritas en todos los casos de transmisión familiar.
Los 6 casos anteriormente documentados poseen, en común, baja estatura, paladar ojival, clinodactilia del quinto dedo, pabellones auriculares prominentes y retraso mental de gravedad variable (tabla 1). El retraso del lenguaje sospechado en el caso índice es una característica bien documentada en los portadores de la deleción del brazo corto de cromosoma 18, a pesar de no siempre se observa. Característicamente, estos individuos tienen dificultades de expresión verbal, con lenguaje expresivo no verbal menos afectado9,10.
Casos de transmisión familiar de la deleción 18p
| Autor | Miembros de la familia | Anomalía cromosómica | Retraso mental | Baja estatura | Aspectos dismórficos |
| Uchida (1965) | Madre | del(18p) | +/− | + | + |
| Hijo | del(18p) | + | + | + | |
| Hija | del(18p) | ? | ? | ? | |
| Velagaleti (1996) | Madre | del(18p11.2) | + | + | − |
| Hija | del(18p11.2) | + | + | + | |
| Tonk (1997) | Madre | del(18p11.2) | + | − | + |
| Feto | del(18p11.2) | ? | ? | ? | |
| Tsukahara (2001) | Madre | del(18p11.2) | +/− | − | + |
| Hija | del(18p11.23) del(18p11.2) | +/− | − | + | |
| Hijo | +/− | − | + | ||
| Rigola (2001) | Madre | del(18p11.3) | + | − | + |
| Feto | del(18p),t(X,Y) | ? | ? | ? | |
| Maranda (2006) | Madre | del(18p11.2) | + | − | + |
| Hija | del(18p11.2) | + | + | + | |
| Hija | del(18p11.2) | +/− | − | + | |
| Caso índice (2007) | Madre | del(18)(11.21) | + | − | + |
| Hijo | del(18)(11.21) | + | + | + |
La madre del caso descrito presenta un cociente intelectual verbal de 64 y revela dificultades de expresión verbal de ideas y la articulación de las palabras.
En todos los casos familiares descritos, la transmisión de la deleción fue materna. Puede tratarse de un sesgo estadístico, teniendo en cuenta el número reducido de casos descritos, o que los portadores masculinos de la deleción tengan alguna alteración de la fertilidad.
Los autores destacan la importancia de la evaluación por los departamentos de genética médica de niños con retraso de crecimiento y malformaciones congénitas.
Debido al riesgo de recurrencia (50%), es importante orientar a esta familia a una consulta de consejo genético, por la posibilidad de diagnóstico prenatal en un futuro embarazo.
