

El síndrome hemofagocítico (SH) se caracteriza por una activación y proliferación incontrolada de histiocitos y linfocitos T, que produce un estado de hipercitocinemia. Hay 2 formas: primaria y secundaria.
ObjetivoAnálisis de los pacientes diagnosticados de SH según los criterios diagnósticos de los protocolos HLH (hemophagocytic lymphohistiocytosis ‘linfohistiocitosis hemofagocítica’)-94 y HLH-2004.
Pacientes y métodosSe revisó de forma retrospectiva la historia clínica de los pacientes diagnosticados de SH, se analizaron los criterios diagnósticos, la forma de presentación, la etiología, el tratamiento administrado y el curso evolutivo.
ResultadosSe diagnosticó a 22 pacientes: 6 con formas familiares, 11 con formas asociadas a infección, 3 con formas asociadas a neoplasia y 2 con síndromes de activación macrofágica (estos pacientes con artritis idiopática juvenil y enfermedad de Crohn [EC]). En el 83,3% de los casos de linfohistiocitosis hemofagocítica familiar (LHF) la edad al diagnóstico fue inferior al año de vida. En un paciente adolescente se diagnosticó una forma primaria de la enfermedad (mutación del gen MUNC13-4). Las manifestaciones clínicas fueron fiebre (100%), hepatoesplenomegalia (85%), adenopatías (31%), palidez (21%), exantema (14%) y alteraciones neurológicas (14%); los hallazgos de laboratorio fueron citopenia (100%), hipertrigliceridemia (93%), hiperferritinemia (86%), elevación de las enzimas hepáticas (78%) e hipofibrinogenemia (40%). Se encontró una reducción de actividad de los linfocitos citolíticos naturales en el 100% de los casos. Se observó hemofagocitosis en la médula ósea en 20 pacientes. En 2 pacientes se realizó una biopsia hepática y ganglionar que demostró hemofagocitosis. Evolución: de los 22 pacientes diagnosticados de SH, 10 pacientes recibieron tratamiento según los protocolos HLH-94 y HLH-2004: 6 con LHF, 3 con formas secundarias al virus de Epstein-Barr y uno a la EC. De éstos, 6 pacientes recibieron un trasplante de progenitores hematopoyéticos (TPH), con evolución favorable en 2 de los casos con LHF. Los otros 12 pacientes con formas secundarias recibieron tratamiento etiológico, con buena evolución en el 83,3%.
ConclusionesLas formas familiares de SH se diagnostican generalmente antes de los 2 años de edad, aunque se presentan formas primarias en edades más avanzadas. El tratamiento quimioterapéutico e inmunosupresor y el TPH constituyen la base del tratamiento de las formas familiares. Las formas secundarias deben recibir tratamiento etiológico y, si la evolución no es favorable, tratamiento quimioterapéutico e inmunosupresor.
Haemophagocytic syndrome (HPS) is a rare syndrome characterised by the uncontrolled activation and proliferation of histiocytes and T cells, leading to a cytokines overproduction. There are two forms of HPS: primary and secondary.
ObjectiveTo analyse patients diagnosed with HPS at the Oncohaematology Department, using HLH-94 and 2004 protocol diagnostic criteria.
Materials and methodsRetrospective study of clinical files of patients diagnosed with HPS, analysing the following features: diagnostic criteria, variability in clinical presentation, aetiology, treatment and outcome.
ResultsTwenty-two patients were diagnosed with HPS: 6 familial haemophagocytic lymphohistiocytosis (FHL), 11 HPS with evidence of infection, 3 HPS associated with malignant disease and 2 macrophage activation syndrome (MAS) in patients with Crohn's disease and Juvenile Idiopathic Arthritis. The onset of FHL was within 1 year of age in 83.3%, except for 1 patient who was adolescent (MUNC13-4 mutations). Symptoms: All patients (100%) had fever at diagnosis, 18 (85%) hepatosplenomegaly, 7 (31%) lymphadenopathy, 5 (21%) pallor, 3 (14%) rash and 3 (14%) neurological symptoms. Laboratory analysis: all patients (100%) had cytopenias at diagnosis, 20 (90.9%) hypertriglyceridaemia, 19 (86%) hyperferritinaemia, 17 (77%) elevated serum liver enzymes, and 9 (40%) hypofibrinogenaemia. Decreased or absent NK-cell activity was detected in all patients (100%). Haemophagocytosis was found more frequently in bone marrow; however, liver or lymph node biopsies were required in two patients to demonstrate this. Outcome: Of the ten patients (6 FHL, 3 Epstein-Barr virus-associated HPS and 1 MAS) treated with HLH-94 and 2004 protocols, six received a stem-cell transplant; of these, 2 with FHL had a favourable outcome. The remaining 12 patients received aetiological/supportive therapy, with complete remission in 83.3%.
ConclusionsThe diagnosis of FHL should be made before the age of 2 years. Advances in genetic studies allow the detection of early and late forms of FHL. Immunochemotherapy and stem-cell transplantation constitute the treatment of FHL and aetiological/supportive therapy of acquired haemophagocytic lymphohistiocytosis, except in severe forms.
Los síndromes hemofagocíticos (SH) se caracterizan por una activación y proliferación no maligna e incontrolada de macrófagos y linfocitos T, asociada a una hiperproducción de citocinas, causante de los principales signos biológicos de este síndrome1,2,3.
Los SH se han clasificado en 2 grupos: primarios o genéticos, y secundarios. En el año 1999 se describió la mutación del gen de la perforina4 (PRF1, 10q21-22), que da lugar a una falta de expresión de la proteína causante de la ausencia de actividad citotóxica de los NK (natural killer ‘linfocitos citolíticos naturales’). Se han observado otras mutaciones de los genes MUNC13-4 (17q25) y STX11 (6q24) asociadas a la linfohistiocitosis hemofagocítica familiar (LHF)5,6,7,8,9,10,11,12,13,14. Algunas inmunodeficiencias, como el síndrome linfoproliferativo ligado al cromosoma X (mutación del gen SH2D1A), pueden manifestarse como un SH.
Los criterios diagnósticos aceptados por el grupo de estudio de la HLH (hemophagocytic lymphohistiocytosis ‘linfohistiocitosis hemofagocítica’) de la Histiocyte Society, publicados por primera vez en 1991 y revisados en 2004, incluyen fiebre elevada persistente, hepatoesplenomegalia, citopenias, hipertrigliceridemia, hiperferritinemia e hipofibrinogenemia. La característica biológica más frecuente es la disminución o ausencia de actividad citotóxica de los NK. El examen histopatológico de la médula ósea muestra histiocitosis importante, con signos de actividad hemofagocítica (figura 1). Estos hallazgos se pueden observar en el bazo, el hígado, los ganglios linfáticos y el líquido cefalorraquídeo15,16. En muchas ocasiones, especialmente en las formas primarias, y sobre todo en las etapas iniciales, puede no identificarse el fenómeno hemofagocítico en la médula ósea.
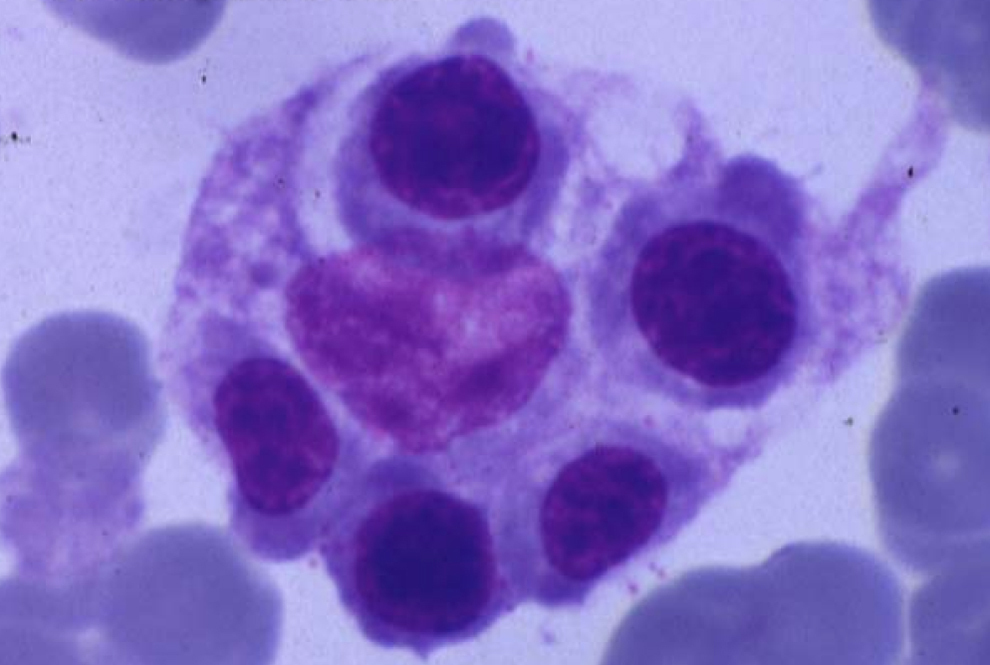
Figura 1. Imagen de hemofagocitosis en la médula ósea.
En las formas primarias, el tratamiento quimioterapéutico (con etopósido) e inmunosupresor (con dexametasona y ciclosporina) asociado a trasplante de progenitores hematopoyéticos (TPH) se ha mostrado útil, como muestran los resultados del protocolo HLH-9417. Sobre la base de éste, en el año 2004 se desarrolló el protocolo HLH-2004 con el objetivo de mejorar los resultados del anterior.
Con el objetivo de conocer en España las formas primarias y secundarias de la enfermedad, se revisó la forma de presentación, la etiología, el tratamiento y la evolución en una serie de 22 pacientes con SH diagnosticados en este centro.
Pacientes y métodosSe revisó la historia clínica de 22 pacientes menores de 18 años de edad, con diagnóstico de SH primario o genético, o secundario sobre la base de los criterios de los protocolos HLH-94 y HLH-2004 (tabla 1). El paciente con síndrome de activación macrofágica (SAM) asociado a artritis idiopática juvenil (AIJ) cumplía los criterios diagnósticos propuestos por Ravelli et al. Estos criterios han sido refrendados recientemente por el Grupo Español de Reumatología Pediátrica18,19,20.
Tabla 1. Criterios diagnósticos de la linfohistiocitosis hemofagocítica (protocolo de 2004 para linfohistiocitosis hemofagocítica)
| 1. Historia familiar o defecto genético conocido 2. Criterios clínicos y de laboratorio (al menos 5 de 8): • Fiebre • Esplenomegalia • Citopenias (⩾2 líneas celulares): Hb<9 g/dl (o<10 g/dl en RN); plaquetas<100×10 9 /l; neutrófilos<1×10 9 /l • Hipertrigliceridemia (⩾265 mg/dl) o hipofibrinogenemia (<1,5 g/l) • Ferritina ⩾500 μg/l • CD25 soluble ⩾2.400 U/ml • Citotoxicidad por células NK disminuida o ausente • Hemofagocitosis en la médula ósea, LCR, bazo o ganglios |
| Apoyan el diagnóstico otros datos como la presencia de sintomatología neurológica con un LCR con moderada pleocitosis o hiperproteinorraquia, elevación de las aminotransferasas e hiperbilirrubinemia, LDH >1.000U/l. El protocolo HLH-94 no incluía ferritina, CD25 soluble y citotoxicidad por células NK como criterios diagnósticos |
Hb: hemoglobina; HLH: hemophagocytic lymphohistiocytosis ‘linfohistiocitosis hemofagocítica’; LCR: líquido cefalorraquídeo; LDH: lactatodeshidrogenasa; NK: natural killer ‘linfocitos citolíticos naturales’; RN: recién nacido.
En todos los pacientes se practicó hemograma, evaluación del funcionalismo hepático y renal e ionograma, coagulación básica, determinación de colesterol, triglicéridos y ferritina, estudio de citotoxicidad de los NK y cuantificación de inmunoglobulinas plasmáticas. En 3 pacientes se realizó el estudio del receptor CD25 soluble, incluido actualmente como criterio diagnóstico de SH en el protocolo HLH-2004.
En todos los casos se practicó aspirado de médula ósea para la demostración de imágenes de hemofagocitosis y estudio de líquido cefalorraquídeo para evaluar la pleocitosis y la hiperproteinorraquia. En pacientes con alta sospecha clínica, en los que no se observaron imágenes de hemofagocitosis en el aspirado de médula ósea, se persiguió el fenómeno hemofagocítico mediante una biopsia hepática o ganglionar.
Se realizaron estudios serológicos o técnicas de reacción en cadena de la polimerasa para el diagnostico vírico (citomegalovirus, virus de Epstein-Barr [VEB], parvovirus B19, virus del herpes tipo 6 y tipo 8, varicela zóster, adenovirus, virus de la inmunodeficiencia humana y virus del herpes simple). Para el estudio de bacterias, hongos y parásitos se emplearon técnicas de cultivo de muestras biológicas.
Se realizaron en todos los pacientes estudios de radiología torácica convencional, ecografía abdominal, tomografía axial computarizada y resonancia magnética craneal.
En todo paciente menor de un año se sospechó una forma familiar. En el protocolo HLH-94, el diagnóstico de las formas familiares se justificaba por la presencia de un antecedente familiar positivo (de primer o segundo grado) y por el cumplimiento de todos los criterios diagnósticos. La existencia de consanguinidad familiar se consideró indicativa de una forma familar de SH. En los últimos años, el estudio de mutaciones genéticas se ha incluido como un criterio diagnóstico en la forma familiar de SH, lo que ha permitido identificar pacientes sin historia familiar previa y en edades más avanzadas.
TratamientoLos pacientes con SH primario o familiar recibieron tratamiento según los protocolos de la Histiocyte Society (utilizados por la Sociedad Española de Hematología y Oncología Pediátrica), que incluyen tratamiento citostásico, inmunosupresor, quimioterapia intratecal en los casos de afectación del sistema nervioso central (SNC), y TPH (tabla 2).
Tabla 2. Esquema general de tratamiento. Protocolos de 1994 y de 2004 para linfohistiocitosis hemofagocítica
| Tratamiento inicial citostásico e inmunosupresor, durante 8 semanas |
| • Forma familiar o genéticamente demostrada: seguir tratamiento citostásico e inmunosupresor, y realizar posterior TPH • Forma no familiar persistente, no demostrada genéticamente: continuar tratamiento citostático e inmunosupresor, y realizar posterior TPH • Forma no familiar resuelta, no demostrada genéticamente: finalizar el tratamiento transcurridas 8 semanas. En caso de reactivación, continuar tratamiento citostático e inmunosupresor, y realizar posterior TPH |
TPH: trasplante de progenitores hematopoyéticos.
Los pacientes con SH secundario recibieron tratamiento etiológico. En los casos de refractariedad a éste, se siguieron pautas de tratamiento similares a las propuestas por la Histiocyte Society para las formas familiares de SH. La valoración clínica, analítica e histológica en la semana 8 del tratamiento permite suspender el tratamiento en los pacientes que han alcanzado la remisión completa y, en el resto de los pacientes, mantener el tratamiento quimioterapéutico e inmunosupresor y el posterior TPH, de la misma manera que en las formas familiares de SH.
ResultadosSe diagnosticó a 22 pacientes de SH, con una razón 1:1 (11 niños, 11 niñas). La mediana de edad al diagnóstico fue de 4 años, con un rango de edad entre 2 meses y 16 años. Cuatro de ellos (19%) presentaban enfermedad previa: enfermedad granulomatosa crónica en 2 casos, enfermedad de Crohn (EC) en uno y AIJ en otro.
DistribuciónSeis pacientes presentaron formas primarias de la enfermedad (27,2%). La edad al diagnóstico fue inferior al año de vida en el 83,3% (5 de 6 casos); se hallaron antecedentes familiares en un caso. En un paciente adolescente, con aplasia medular por parvovirus B19, se demostró una LHF tipo 3 (MUNC13-4). En 3 pacientes con forma familiar de SH se detectó infección vírica asociada a VEB, parvovirus B19 y virus del herpes tipo 6.
Dieciséis pacientes (72,7%) presentaron SH secundarios: SH asociados a infección (SHAI) en 11 casos (68,7%), a neoplasia en 3 casos (18,7%) y a enfermedad autoinmunitaria en 2 casos (12,6%). De los 11 casos de SHAI, 5 fueron secundarios a infección por VEB y 4 a infección por Leishmania (tabla 3).
Tabla 3. Distribución etiológica de los síndromes hemofagocíticos secundarios
| SH asociado a infección (11 de 16; 68,7%) | Virus de Ebstein-Barr: 5 |
| Virus del herpes tipo 6: 1 | |
| Leishmania: 4 | |
| Salmonella: 1 | |
| SH asociado a neoplasia (3 de 16; 18,7%) | Leucemia aguda linfoblástica |
| Leucemia o linfoma T | |
| Rabdomiosarcoma | |
| SAM (2 de 16; 12,6%) | Artritis crónica juvenil |
| Enfermedad de Crohn |
SAM: síndrome de activación macrofágica; SH: síndrome hemofagocítico.
Se detectaron mutaciones en el 50% de las formas primarias: del gen MUNC13-4 en 2 pacientes, correspondiente a la forma familiar tipo 3; y del gen SH2D1A en el cromosoma X, en 1 paciente con síndrome linfoproliferativo ligado al cromosoma X.
Presentación clínicaLa fiebre prolongada fue el motivo de consulta más frecuente, presente en 14 pacientes (63,6%). Se observó fiebre en 22 casos (100%), hepatoesplenomegalia en 18 casos (81,8%), linfadenopatías en 7 casos (31,8%), exantema cutáneo en 3 casos (13,6%) y alteraciones neurológicas en 3 casos (13,6%). Los hallazgos de laboratorio más comunes fueron citopenias (2 o 3 líneas celulares) en todos ellos, hipertrigliceridemia en 20 pacientes (90,9%), hiperferritinemia en 19 pacientes (86,3%), elevación de las aminotransferasas en 17 pacientes (77,2%), hipofibrinogenemia en 9 pacientes (40,9%) y ausencia o disminución de la actividad citotóxica de NK en 22 pacientes (100%). En 20 pacientes (90,9%) se observaron imágenes de hemofagocitosis en la médula ósea, y se demostraron mediante biopsia ganglionar o hepática en otros 2 pacientes. En los 4 casos de hemofagocitosis asociada a infección por Leishmania, el estudio de médula ósea aportó el diagnóstico etiológico. Cinco pacientes (22,7%) presentaron pleocitosis e hiperproteinorraquia en líquido cefalorraquídeo: 3 con formas primarias y 2 con secundarias (VEB y EC); se observaron en la resonancia magnética craneal de todos ellos lesiones multifocales en el cerebelo y en el parénquima cerebral.
Tratamiento y curso evolutivoFormas familiaresSeis pacientes recibieron tratamiento según los protocolos HLH-94 y HLH-2004. Cinco recibieron TPH. Con un seguimiento de 3 años, la evolución ha sido favorable en 2 de ellos (40%). En el paciente afectado de un síndrome linfoproliferativo ligado al cromosoma X se desarrolló una infección fúngica invasiva durante la fase de tratamiento quimioterapéutico e inmunosupresor, y falleció.
La reactivación sistémica y en el SNC descrita en las formas familiares de SH se observó en un caso (paciente 5).
Formas secundariasEn el contexto de las formas secundarias de SH, 12 pacientes recibieron sólo tratamiento etiológico: 8 pacientes con formas asociadas a infección (4 por Leishmania, 2 por VEB, uno por Salmonella y 1 por virus del herpes tipo 6), 3 con formas asociadas a neoplasia (linfoma, leucemia y rabdomiosarcoma) y 1 con SAM. En este grupo, 2 pacientes fallecieron (16,6%): un paciente con enfermedad granulomatosa crónica y leishmaniasis visceral falleció por infección fúngica invasiva, y otro con linfoma T refractario a tratamiento citostásico. Los 4 pacientes afectados de formas graves recibieron tratamiento etiológico combinado con quimioterapia y tratamiento inmunosupresor: 3 con formas asociadas a infección por VEB y 1 con forma asociada a EC. Un paciente con infección por VEB presentó respuesta clínica, analítica e histológica a las 8 semanas, finalizó su tratamiento y se halla en remisión completa a los 2 años. Los otros 2 pacientes con VEB fallecieron: 1 por disfunción multiorgánica a las 48h del diagnóstico, y el otro por progresión de la enfermedad tras el TPH. El paciente con EC y SAM falleció por disfunción multiorgánica.
Las tabla 4,tabla 5 resumen los tratamientos realizados en las formas de SH primario y secundario.
Tabla 4. Tratamiento de los síndromes hemofagocíticos primarios *
| N° | Sexo | Genética | Infección al dx. | Tratamiento | TPH | Evolución |
| 1 | M | No estudiado | No | Protocolo HLH-94 | SCU | Fallecimiento por progresión |
| 2 | F | No estudiado | No | Protocolo HLH-94 | Familiar con una diferencia | Fallecimiento por MRT |
| 3 | M | MUNC13-4 | Parvovirus B19 | Protocolo HLH-04 | FI | Fallecimiento por MRT |
| 4 | F | No identificada | No | Protocolo HLH-04 | FI | Viva a los 3 años |
| 5 | F | MUNC13-4 | Virus del herpes tipo 6 | Protocolo HLH-04 | SCU | Viva a los 3 años |
| 6 | M | SAP/SH2D1A | VEB | Protocolo HLH-04 | – | Fallecimiento por IFI |
dx: infección al diagnóstico; F: femenino; FI: familiar idéntico; HLH: hemophagocytic lymphohistiocytosis ‘linfohistiocitosis hemofagocítica’; IFI: infección fúngica invasiva; M: masculino; MRT: mortalidad relacionada con el trasplante; SCU: sangre de cordón umbilical; TPH: trasplante de progenitores hematopoyéticos; VEB: virus de Epstein-Barr.
* Regímenes de acondicionamiento según los protocolos de 1994 y de 2004 para linfohistiocitosis hemofagocítica.
Tabla 5. Tratamiento de los síndromes hemofagocíticos secundarios
| N.° | Sexo | Etiología | Historia previa | Tratamiento | Evolución |
| 1 | F | VEB | No | Protocolo HLH-94 y TPH haploidéntico | Fallecimiento por progresión |
| 2 | F | Linfoma T | No | Quimoterapia | Fallecimiento por progresión |
| 3 | M | Leishmania | No | Anfotericina B | Vivo |
| 4 | M | Leishmania | No | Anfotericina B | Vivo |
| 5 | F | VEB | No | Ganciclovir y GGIV | Viva |
| 6 | M | LAL | No | Quimioterapia | Vivo |
| 7 | F | VEB | No | Ganciclovir y GGIV | Viva |
| 8 | M | Virus del herpes tipo 6 | No | Foscarnet y GGIV | Viva |
| 9 | M | ACJ | IS | Corticoides y GGIV | Vivo, TPH Autólogo |
| 10 | M | Leishmania | EGC | Anfotericina B | Fallecimiento por IFI |
| 11 | M | Leishmania | EGC | Anfotericina B y otros | Vivo |
| 12 | M | VEB | No | Protocolo HLH-04, durante 8 semanas | Vivo, sin seguimiento |
| 13 | F | Enfermedad de Crohn | IS | Protocolo HLH-04 | Fallecimiento por FM |
| 14 | F | VEB | No | Protocolo HLH-04 * | Fallecimiento por FM |
| 15 | F | Rabdomiosarcoma | No | Quimioterapia y Corticoides | Vivo |
| 16 | F | Salmonella | No | Cefalosporina | Vivo |
ACJ: artritis crónica juvenil; EGC: enfermedad granulomatosa crónica; F: femenino; FM: fallo multiorgánico; GGIV: gammaglobulina inespecífica intravenosa; HLH: hemophagocytic lymphohistiocytosis ‘linfohistiocitosis hemofagocítica’; IFI: infección fúngica invasiva; IS: inmunosupresión; LAL: leucemia aguda linfoblástica; M: masculino; VEB: virus de Epstein-Barr.
* Recibió una dosis de etopósido y corticoterapia. Falleció a las 48h.
Trece pacientes están libres de enfermedad con una mediana de seguimiento de 86 meses (rango entre 24 y 118 meses): 2 con formas familiares, 8 con formas asociadas a infección (3 con VEB, 3 con leishmaniasis, 1 con virus de herpes tipo 6 y 1 con Salmonella), 2 con formas asociadas a neoplasia (leucemia aguda linfoblástica y rabdomiosarcoma) y 1 con SAM (artritis crónica juvenil).
DiscusiónEl SH es el resultado de una alteración genética o adquirida en la regulación de la activación macrofágica: se produce una liberación de citocinas proinflamatorias que pueden condicionar un daño tisular irreversible que puede ser mortal1,2,3.
Se distinguen 2 formas: la primaria o genética, y la secundaria. Las formas primarias se subdividen en LHF y en síndromes de inmunodeficiencia, como el síndrome de Griscelli, el síndrome de Chédiak-Higashi y el síndrome linfoproliferativo ligado al cromosoma X, que pueden desarrollar SH de forma esporádica, aunque frecuente. La LHF se hereda con carácter autosómico recesivo y presenta una incidencia estimada de 1:50.000 niños. El 90% de los pacientes se diagnostican por debajo del año de vida, pero se han descrito algunos casos en adolescentes y en adultos jóvenes, como sucede en esta serie. Se encuentran alteraciones genéticas en el 50 al 60% de los casos. En los pacientes con SH primario aquí presentados se identificó la mutación del gen MUNC13-4 en 2 de ellos (uno era un adolescente), y la mutación del gen SH2D1A en un niño con un síndrome linfoproliferativo ligado al cromosoma X. En 1 paciente no se identificó ninguna de las mutaciones conocidas, y en los otros 2 no se realizó estudio genético4,5,6,7,8,9,10,11,12,13,14,15,16,17.
Los procesos infecciosos pueden actuar como desencadenante en un SH primario, y puede ser difícil la distinción de las formas de SH secundario. Se recomienda el estudio de la mutación SH2D1A en todo paciente varón con hemofagocitosis e infección asociada a VEB, y también en varones con sospecha clínica, aunque no se demuestre la infección por VEB. Esta mutación se demostró en el caso 6.
Dentro de las formas de SH secundario destacan el SHAI, el SH asociado a neoplasia y el SAM o asociado a enfermedades autoinmunitarias. El SHAI representa del 50 al 60% de las formas secundarias y, de éstas, el 50% están representadas por infecciones víricas, fundamentalmente por VEB. Otro grupo importante lo constituye la infección por Leishmania21,22,23,24. De la misma forma, esta distribución se reproduce en este grupo de pacientes.
La patogenia del SH viene determinada por la excesiva activación de linfocitos y de macrófagos, que condiciona un síndrome hiperinflamatorio por la masiva liberación de citocinas, como el factor de necrosis tumoral alfa, la interleucina (IL)-1, IL-6, IL-8, IL-10, IL-12, IL-18, el interferón γ y la proteína macrofágica inflamatoria 1-alfa (MIP 1-alfa). La hiperproducción de citocinas es la causante de los 3 grandes signos clínicos del SH: la fiebre, las citopenias y la inflamación25. La fiebre es el signo de consulta más frecuente, aunque hay otras formas de presentación atípicas, que pueden dificultar el diagnóstico26,27,28,29.
Un estudio negativo en médula ósea no excluye el SH. La hemofagocitosis puede no estar presente al diagnóstico y puede ser necesaria la práctica reiterada de aspirados medulares, como sucedió en 3 de 20 (15%) de estos pacientes. En ocasiones, la práctica de una biopsia hepática o ganglionar puede ayudar, como ocurrió en 2 de los 22 (9%) pacientes. Sin embargo, la demostración del fenómeno de hemofagocitosis no es un requisito indispensable para establecer el diagnóstico de SH, siempre que se cumplan los otros criterios clínicos.
El objetivo fundamental en el tratamiento del SH es la supresión de la excesiva respuesta inflamatoria, que puede poner en peligro la vida del paciente. Anteriormente a la introducción de los protocolos HLH-94 y HLH-2004, la supervivencia de las formas primarias de SH era muy escasa, cercana al 5%. La incorporación de agentes inmunosupresores y quimioterapéuticos, como la dexametasona, la ciclosporina y el etopósido, asociados al TPH en las formas de SH primario, ha logrado supervivencias globales a los 3 años cercanas a entre el 50 y el 70% de los casos30,31,32,33,34. En los pacientes con formas secundarias de SH, el tratamiento etiológico y de soporte es el de elección; y se reserva el tratamiento quimioterapéutico e inmunosupresor para los casos graves o refractarios.
Los pacientes pueden presentar afectación del SNC, que puede dejar secuelas. En ocasiones, también es posible la reactivación sistémica o en el SNC durante el tratamiento y se precisa de una intensificación de éste.
Merecen especial atención, el SH asociado a VEB y el SH por Leishmania. En las formas graves de SHAI por VEB, la incorporación al tratamiento de etopósido dentro de las primeras 4 semanas del diagnóstico se ha mostrado como la única variable con impacto pronóstico, y ha mejorado de una forma notable las tasas de supervivencia. Imashuku et al han intentado estratificar a los pacientes con SH asociado a VEB en 2 grupos: de bajo y de alto riesgo, con la idea de poder diferenciar su tratamiento35,36. Recientemente, se ha descrito el uso de quimioterapia y rituximab (anticuerpo monoclonal anti-CD20) en el SH asociado a VEB. En esta serie, 5 pacientes presentaron un SH asociado a VEB, y la mortalidad global fue del 40% (2 de 5 casos). En el SHAI por Leishmania, la utilización de anfotericina B suele ser suficiente para su resolución37. En este grupo, los 4 casos recibieron tratamiento con anfotericina B, se obtuvo una respuesta completa en 3, y falleció 1 paciente afectado de enfermedad granulomatosa crónica por una infección fúngica invasiva.
Es importante el reconocimiento precoz del SH y su tratamiento inmediato. La existencia de unos criterios diagnósticos y terapéuticos, definidos por el grupo de estudio de la HLH de la Histiocyte Society, permite iniciarlo38. El estudio genético de las formas primarias permitirá identificar adolescentes o adultos jóvenes que presenten formas familiares de SH.
