


El síndrome de Fraser (SF; OMIM 219000) es una enfermedad poco frecuente, de herencia autosómica recesiva, caracterizada por una expresión variable de múltiples malformaciones congénitas, como criptoftalmos (párpados fusionados o ausentes), sindactilia cutánea, genitales ambiguos, labio leporino, fisura palatina, agenesia renal, hernias umbilicales y malformaciones anorrectales, cardiacas o esqueléticas.
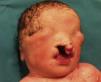
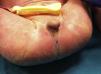
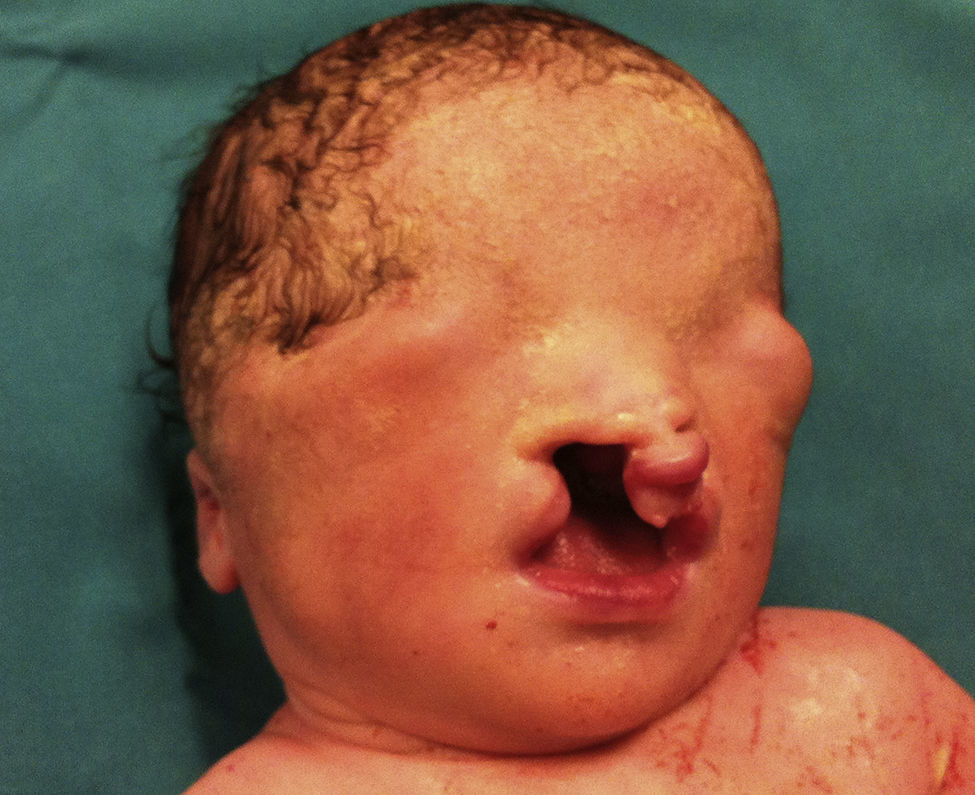
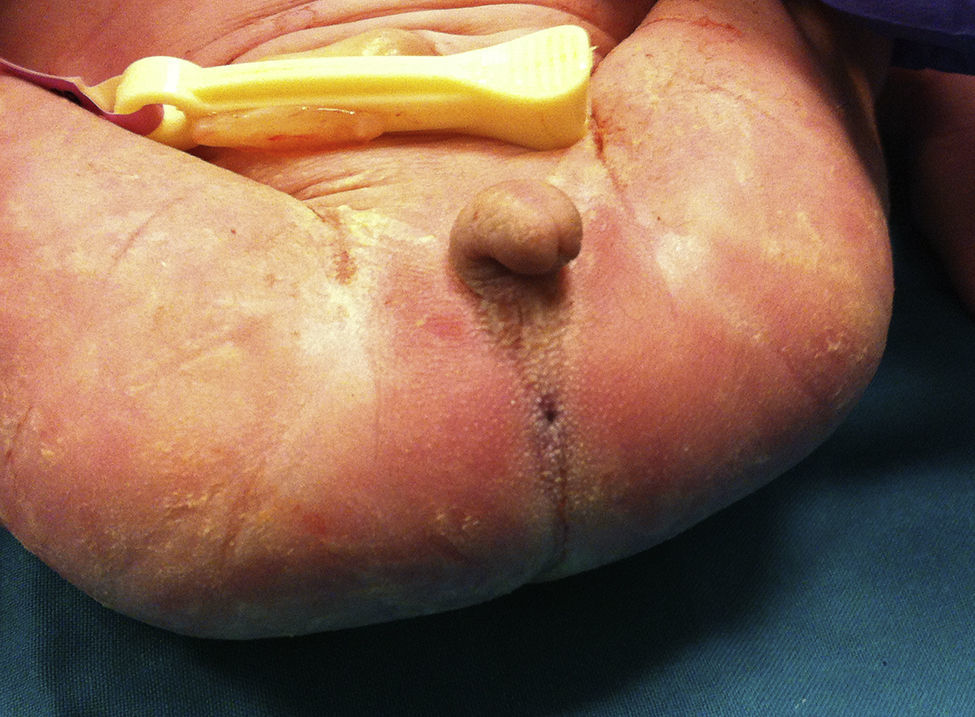
Presentamos a un neonato, fruto de una séptima gestación, de una madre de 26 años y un padre de 40 años, ambos de origen marroquí, primos hermanos. Los 6 embarazos anteriores en Marruecos terminaron con 3 abortos espontáneos, 2 mortinatos malformados y una niña normal, sin que fuera posible disponer de más datos. Las ecografías desde la semana 20 de gestación mostraron oligoamnios, pulmones ecogénicos, hernia umbilical y genitales ambiguos. Las ecografías cardiaca y renal fueron normales. Nació una niña prematura tardía con cariotipo 46XX, de 34 semanas, 1.930g, con múltiples malformaciones, como criptoftalmos bilateral, labio leporino, fisura palatina (fig. 1), sindactilias en manos y pies, genitales ambiguos (fig. 2) e imperforación anal, que falleció a los 10 min de vida. Los padres no permitieron la autopsia pero sí la toma de muestras. Ante la sospecha de SF, se realizó estudio genético completo del gen FRAS1, siendo normal, y de los exones e intrones 1,3-4, 9,12 y 16-24 del gen FREM2 (GenBank número de acceso: NM_207361.4) usando el analizador Applied Biosystems 3500 OX (Foster City, California, EE. UU.), encontrándose una deleción en homocigosis c.4733de1A (p.H1578LfsX20) en el exón 1 del gen FREM2, apareciendo un codón de terminación prematuro formándose una proteína truncada con 1.596 aminoácidos frente a los 3.169 de la proteína normal. Esta alteración no ha sido descrita según nuestro conocimiento como causante de SF.
Comentario: en una serie europea reciente1, la prevalencia estimada de SF era de 0,2 casos por 100.000 o 1/495.633 partos, aunque algunos podrían no estar diagnosticados, especialmente en mortinatos. El 82% de los casos con diagnóstico prenatal condujo a una terminación voluntaria del embarazo. Hay más de 250 casos publicados de pacientes con SF. Los criterios diagnósticos más recientes2,3 incluyen criterios mayores (sindactilia cutánea, criptoftalmos, genitales ambiguos, alteraciones urinarias y respiratorias, historia familiar) y criterios menores (malformaciones nasales, del canal auditivo, anorrectales, hernia umbilical, defectos de la osificación craneal). El diagnóstico clínico puede hacerse por la presencia de 3 criterios mayores, 2 mayores y uno menor, o un criterio mayor y 3 menores. En los casos familiares, el diagnóstico prenatal puede hacerse por ultrasonografía, siendo el oligoamnios, la no visualización de los riñones y la hiperecogenicidad pulmonar los hallazgos más frecuentes, indicando su existencia la recurrencia del SF4.
El SF es una condición genéticamente heterogénea. Se han identificado mutaciones en el gen FRAS1 (cromosoma 4q21) y FREM2 (cromosoma 13q13). Ambos genes codifican proteínas de la matriz extracelular esenciales para la adhesión entre la membrana basal epidérmica y el tejido conectivo durante el periodo de la embriogénesis. Recientemente5,6, se han descrito mutaciones en el gen GRIP1 (cromosoma 12q14) que codifica una proteína que interactúa con las proteínas anteriores, como causantes del SF. Mientras que se han descrito varias mutaciones en el gen FRAS17, solamente se han comunicado 2 mutaciones en el gen FREM2 hasta la fecha8–10. Actualmente, se está estudiando el estado de portadores de la mutación en los padres.
Presentamos los hallazgos moleculares de un recién nacido con SF, en el que el estudio reveló una mutación no descrita en el gen FREM2. Este hallazgo expande el espectro de las mutaciones causantes del SF y la posibilidad de ampliar el diagnóstico para los pacientes y sus familias.
