



La craneosinostosis consiste en una fusión patológica precoz de una o varias suturas craneales. El 20% de los casos corresponde a formas sindrómicas con patrones hereditarios mendelianos, mientras que el 80% restante a formas no sindrómicas, pero con transmisión hereditaria en el 10-14% de los casos. A propósito de 2 pacientes con síndrome de Crouzon, se revisan los aspectos clínicos y genéticos.
Pacientes y métodosPaciente 1: niña de 35 días con macrocefalia progresiva, abombamiento de la fontanela, proptosis ocular, hipertelorismo y estrabismo divergente. Rx de cráneo con sinostosis de la sutura sagital. Fue intervenida quirúrgicamente a los 3 y 8 meses por desarrollo de pansinostosis. Paciente 2: niño de 3 años 8 meses con cefaleas de tipo migrañoso de un año de evolución. Presentaba acantosis nigricans. Rx de cráneo y TC craneal con impresiones digitales y fondo de ojo con discreto borramiento papilar. Tras 18 meses apareció edema de papila y en la TC craneal se detectó pansinostosis, requiriendo intervención quirúrgica.
ResultadosHemos presentado un paciente con síndrome de Crouzon clásico (paciente 1) y otro con acantosis nigricans (paciente 2), diagnosticándose por su particular fenotipo clínico.
ConclusionesDada la amplia diversidad de formas alélicas en los genes FGFR que cursan con craneosinostosis, conociéndose hasta 10 entidades, realizamos una revisión de las mismas. En las formas sindrómicas, como nuestros 2 casos, conviene detallar al máximo los signos clínicos pues pueden orientar el diagnóstico, y el estudio molecular permitirá en ocasiones confirmarlo y ofrecer asesoramiento genético a las familias.
Craniosynostosis is an abnormal and premature fusion of any cranial suture. Twenty per cent of them involve any specific syndrome with Mendelian transmission; the other 80% are “non syndromic”, although but 10-14% of them are genetically transmitted. Using the experience of two patients with Crouzon syndrome, a clinical and genetic review is performed.
Patients and methodsPatient 1: girl of 35 days of age with progressive macrocephaly, protrusion of fontanel, ocular proptosis, hypertelorism and divergent strabismus. Cranial RX with sagittal synostosis. Surgical operation was performed with 3 months and 8 months of age due to development of pansynostosis. Patient 2: boy of 3 years 8 months of age with headaches of migrainous type of one year onset. He had acanthosis nigricans. Cranial RX and cerebral CT with evident digital markings and fundus of eye with undefined papillary limits, but 18 month later oedematous papilla were evident and pansynostosis was detected, so surgery was performed.
ResultsWe present a patient with classical Crouzon syndrome (patient 1) and another with acanthosis nigricans (patient 2), both diagnosed by the description of characteristic clinical features.
ConclusionsTen craniosynostotic clinical forms are currently known as allelic variations of the FGFR genes, and as such have reviewed them. As in our two cases, in syndromic types is very important the accurate study of the phenotype to orientate the diagnosis, although the molecular study will confirm it in many patients and genetic counselling offered.
La craneosinostosis consiste en una fusión patológica precoz de una o varias suturas craneales. El cráneo se deforma creciendo en sentido paralelo a la sutura afectada, intentando compensar el crecimiento hacia las suturas abiertas. La incidencia en la población general de todas las craneosinostosis es de 1 por cada 2.000 a 2.500 recién nacidos vivos. El 20% de los casos forman parte de síndromes complejos (se conocen más de 150), siendo su herencia de tipo mendeliana. Pero entre un 10 y 14% de las craneosinostosis no sindrómicas también existe transmisión hereditaria, conociéndose en la actualidad cuatro genes responsables: a) TWIST1 (7p21), causante de la craneosinostosis aislada tipo 11; b) MSX2 (5q34-q35), causante de la craneosinostosis aislada tipo 2 (MIM 123101); c) FGFR2 (mutación M943T) (10q25-q26)2 (MIM 176943), y d) FGFR3 (4p16.3)2,3. Estos 4 genes también son responsables de formas sindrómicas.
En este trabajo se presentan 2 pacientes no emparentados afectados por el síndrome de Crouzon, diagnosticados por su fenotipo clínico. En este síndrome pueden hallarse mutaciones en los genes FGFR (fibroblast growth factor receptor) 2 o FGFR3. Sin embargo, existen hasta 10 formas alélicas de los genes FGFR que manifiestan craneosinostosis y, aunque cada entidad tiene un fenotipo bastante particular, algunos de los signos clínicos son superponibles y con expresividad variable4 pudiendo resultar difícil la clasificación de algunos pacientes. Para simplificarlo, en la tabla 1 se esquematizan los signos clínicos más característicos de cada entidad, haciéndose también referencia a los genes responsables de cada una y al porcentaje actual de detección de las mutaciones2.
Formas alélicas de los genes FGFR1, FGFR2 y FGFR3 que cursan con craneosinostosis
| 1. Craneos. cor. | 2. S. Crouzon | 3. S. Apert | 4. S. Pfeiffer | 5. S. Jackson-Weiss | 6. S. Saethre-Chotzen | 7. S. Antley-Bixler | 8. S. Beare-Stevenson | 9. S. Crouzon con ac. nigric. | 10. S. Muenke | |
| Gen/mutación | TWIST1, MSX2, FGFR2, FGFR3. | FGFR2 | FGFR2 | FGFR2 (tipo I, II, III) FGFR1 (5% tipo I) | FGFR2 FGFR1 (un caso) | TWIST1 FGFR2 (2 famil.) | FGFR2 (AD) POR (AR) | FGFR2 (mayoría) FGFR3 | FGFR3 (G1172A) | FGFR3 (P250R) |
| Detec. mut.2 | 100% en FGFR2 y FGFR 3 | > 50% | > 98% | 67% en cada tipo | % desconocido | 75% en TWIST1 | % desconocido | % desconocido | 100% | 100% |
| Herencia | AD | AD | AD | AD | AD | AD | AD (FGFR2) | Desconocida | AD | AD |
| Dismorfia craneo-facial | Craneal | Sí | Sí | Sí | Sí | Ptosis palpebr., hipertelorismo | Cráneo forma trapezoide | Sí | Sinostosis solo coronal | Sí |
| Trigonocefalia | No | Raramente (7%) | Raramente | Posible | No | No | No | Posible | No | Posible |
| «Lagunas» parietales | No | No | No | No | No | Sí | Sí | No | No | No |
| Manos | Nl | Posibles anomalías Rx | Sindactilia cutánea (posible ósea) | Braquidactilia, sindactilia 2-3 | Nl la mayoría | Braquidactilia, sindactilia 2-3 | Camptodactilia, aracnodactilia | Nl | Posible leve acortam. falang. | Braquidactilia, posible fusión en carpo |
| Pulgares | Nl | Nl | Cortos (ocasional sindactilia) | Anchos, cortos (desviación medial) | Nl | Anchos | Nl | Nl | Nl | Posibles anomalías falanges |
| Polidactilia | No | No | Posible en manos y pies | Falange distal bífida | No | No | No | No | No | No |
| Antebrazos | Nl | Posible sublux. cabeza radio | Posible sinostosis radio-humeral | Sinostosis radio-cubital | Nl | Nl | Curvados, sinostosis radio-cubital | Nl | Nl | Nl |
| Codos | Nl | Posible anquilosis | Posible limitac. extensión | Sinostosis radio-humeral | Nl | Posible limitac. extensión | Sinostosis radio-humeral | Nl | Nl | Nl |
| Pies | Nl | Nl | Sindactilia cutánea (posible ósea) | Cortos (posible sinsactilia) | Sinost. tarsiana, sindac. cut. 2-3. 1.er metat. corto | Sindactilia 3-4 | Posibles zambos | Nl | Nl | Posible fusión tarso |
| 1.er dedo pie | Nl | Nl | Separados (ocasional sindactilia) | Anchos y cortos, hallux varus | Anchos, hallux varus | Anchos, hallux valgus | Nl | Nl | Nl | Posible ancho |
| Cardíaco, renal | Nl | Nl | Posible afectación | Nl | Nl | Posible afectación | Posible afectación | Posible afectación | Posible afectación | Nl |
| SNC | Nl | Hidrocef. (47%), R. ment. (12%) | Posibles hidrocefalia, ACC y Chiari I | Posible hidrocefalia | Nl | Intelecto en general Nl | Posible normalidad | Posible Chiari e hidrocefalia | Posible hidrocefalia o Chiari I | R. ment. (50%) (pos. megalen- cefalia aislada) |
| Piel | Nl | Nl | Nl | Nl | Nl | Nl | Nl | Cutis girata, acantosis nigricans | Acantosis nigricans | Nl |
| Sordera | No | Posible esten. canal auditivo | Posible | Posible | No | Posible | Sí; displasia auricular | Posible displasia auricular | No | Si (neuro-sens. o mixta) |
| Otros | Chiari I (73%), fus. vert. (18%) | Fus. vert. (68%) | Muy amplio espectro clínico | Fémures curv. Posib. genitales ambiguos | Posible anomal. genital, dental y hernia umbilic. | Pos. atr. coanas, fisura palatina | Muy amplio espectro clínico |
ac. nigric.: acantosis nigricans; ACC: agenesia del cuerpo calloso; acortam. falang.: acortamiento de falanges; AD: autosómica dominante; AR: autosómica recesiva; Craneos. cor.: craneosinostosis coronal; fémures curv.: fémures curvados; fus. vert.: fusiones vertebrales; Nl: normal; pos.: posible; R. ment.: retraso mental.
En el Servicio de Pediatría del Consorci Sanitari de Terrassa, desde 1990 hasta la actualidad hemos diagnosticado un total de 12 pacientes con craneosinostosis: 8 formas aisladas y 4 sindrómicas (un síndrome de Apert, un síndrome de Baller-Gerold y 2 síndromes de Crouzon). Estos 2 últimos, diagnosticados en 2008 y 2009, fueron derivados para tratamiento quirúrgico al Servicio de Neurocirugía del Hospital Sant Joan de Déu (Esplugues de Llobregat) y son el objeto de esta revisión.
ResultadosPaciente 1Niña de 35 días de vida, remitida por presentar abombamiento de la fontanela durante las últimas semanas con aumento del perímetro craneal, pasando de +2 DE de recién nacida hasta +3 DE. Antecedentes familiares: primer hijo; padres con morfología y perímetro craneal normales; un aborto espontáneo anterior. Antecedentes personales: embarazo con diabetes gestacional y parto a las 40 semanas mediante cesárea. Test de Apgar 9-10. Peso: 3.410 g (pc 40), longitud: 49,5cm (pc 40), perímetro cefálico: 36,5cm (pc 98).
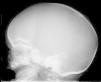
En la exploración destacaba un perímetro cefálico de 39,3cm (+3 DE), con frente prominente, moderada dolicocefalia, marcado hipertelorismo ocular con proptosis y estrabismo divergente. Manos y pies, normales. Fontanela ligeramente abombada, palpándose un ribete en la sutura sagital. Su fenotipo facial concordaba con un síndrome de Crouzon. Rx craneal con sinostosis sagital (fig. 1). Fondo de ojo normal. Precisó intervención quirúrgica a los 3 y 8 meses por agravamiento evolutivo de su morfología cráneo-facial y desarrollo de pansinostosis. La segunda intervención consistió en un avance fronto-orbitario. Tras 21 meses de seguimiento, mantiene normalidad en su desarrollo psicomotor con mejoría evidente de su fenotipo craneo-facial. Mediante secuenciación de los exones 8 y 10 del gen FGFR2 y del exón 6 del gen FGFR3 no se encontraron mutaciones.
Paciente 2
Niño de 3 años y 8 meses de edad, remitido para estudio de cefaleas intermitentes de tipo migrañoso, de un año de evolución. Antecedentes familiares: padres y hermana con morfología y perímetro craneal normales. Antecedentes personales: embarazo y parto normales. Peso: 3.100 g (pc 25), longitud: 49cm (pc 25), perímetro cefálico: 35,5cm (pc 60). Bronquitis obstructivas de lactante. Obesidad desde los 12 meses. Deambuló a los 2 años. Leves dificultades de pronunciación. Intelecto normal.
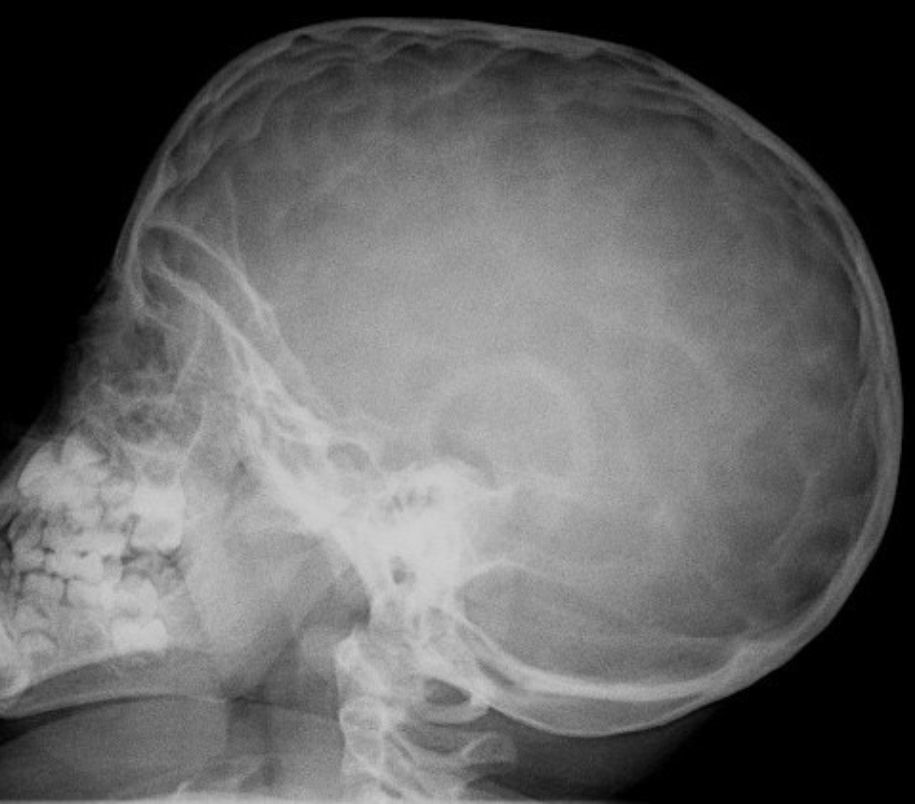
En la exploración destacaba obesidad, con un peso de 29kg (+7 DE), talla de 104,5cm (+1,5 DE) y perímetro craneal de 50,6cm (0 DE). Presentaba una leve proptosis ocular y en el fondo de ojo existía un discreto borramiento de los bordes papilares. En la Unidad de Endocrinología se detectó acantosis nigricans en el cuello y las axilas, manifestada a partir de los 2 años (fig. 2). Rx de cráneo y TC cerebral con evidentes impresiones digitales (fig. 3). El fenotipo cráneo-facial y la anomalía dermatológica hicieron sospechar un síndrome de Crouzon con acantosis nigricans. Los potenciales evocados visuales y la RM cerebral fueron normales. Transcurridos 18 meses, apareció edema de papila en el fondo de ojo y la TC craneal detectó pansinostosis, incluida levemente la metópica. Se intervino quirúrgicamente realizándose un avance fronto-supraorbitario, respetando las órbitas dado que no eran muy pequeñas, junto a una fragmentación biparietal, consiguiéndose aumentar el volumen intracraneal. Tras 2 años se mantiene asintomático y se ha reducido la proptosis ocular. No ha sido posible el estudio genético por haberse trasladado a otra comunidad autónoma.
Discusión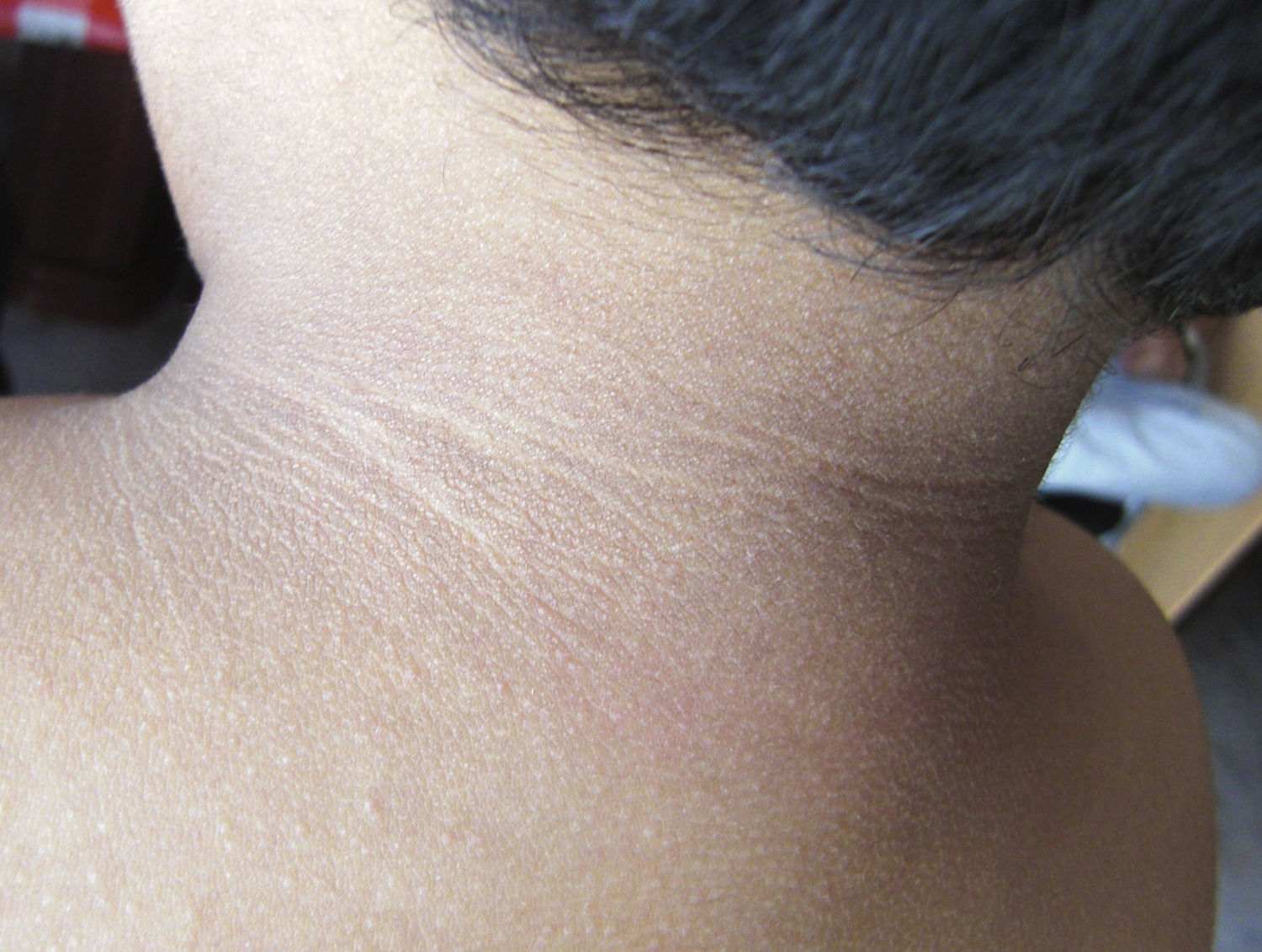
Estos 2 pacientes presentan 2 formas diferentes de síndrome de Crouzon, el primero atribuido a una mutación en el gen FGFR2 (detectándose la misma al menos en la mitad de los pacientes2, sin hallarla en nuestro caso) y el segundo en FGFR3 (una mutación recurrente, aunque no la hemos podido estudiar en el nuestro).
Si el estudio del gen FGFR2 en un paciente con sospecha de síndrome de Crouzon se realiza antes de los 2-3 años, las probabilidades de obtener un resultado negativo serán mayores que si se realiza a una edad posterior, por la posibilidad de se trate de un síndrome de Crouzon con acantosis nigricans sin que todavía se haya manifestado la anomalía cutánea, localizándose la mutación en FGFR3.
Crouzon describió en 1992 a una madre y su hija afectadas por una craneosinostosis que abarcaba diversas suturas, provocando una deformidad cráneo-facial junto a exoftalmos. Posteriores publicaciones médicas de casos similares configurarían este síndrome, recibiendo el nombre del primer autor. Su herencia es AD (autosómica dominante), con penetrancia completa y expresividad variable. La mitad de los casos son debidos a mutacions de novo. Se estima una incidencia de 1 por cada 60.000 recién nacidos vivos y constituye el 4,8% de todas las craneosinostosis. No tiene predominio racial ni de sexo.
La craneosinostosis suele detectarse durante el primer año de vida y puede abarcar la bóveda craneal, base de cráneo, órbitas y complejo maxilar. La precocidad y el orden de afectación de las suturas pueden variar, existiendo una cierta diversidad en su espectro clínico. Destaca principalmente hipertelorismo con exoftalmos (por órbitas poco profundas), estrabismo divergente, nariz en pico curvado, labio superior fino, hipoplasia mediofacial (por maxilar superior reducido) y un relativo prognatismo. En raras ocasiones también pueden existir anomalías oculares. Un 7% de los casos son muy graves4, adquiriendo el cráneo una forma trilobulada (también llamada trigonocefalia, en «hoja de trébol», «clover-leaf» o «kleeblattschädel»). Si se demora la cirugía de estos casos, la hipertensión intracraneal y el edema de papila provocan grave déficit visual. Un 47% también presenta hidrocefalia progresiva, con posible herniación amigdalar y/o siringomielia. Algunos centros tratan la hidrocefalia mediante ventriculostomía endoscópica en el tercer ventrículo, evitando la colocación de una válvula derivativa en más de la mitad de los casos5. El 12% de los casos presentan retraso cognitivo.
Existe un espectro clínico muy amplio para este síndrome, cuya forma de presentación más grave es neonatal. En ocasiones, el estudio clínico familiar puede desvelar casos muy leves en el adulto, que no requieren ningún tratamiento. En los casos severos la cirugía debe ser precoz, anticipándose a posibles complicaciones, especialmente las del nervio óptico6. Una de las técnicas consiste en realizar un avance fronto-orbitario, que evitará la hipertensión intracraneal, aparte de mejorar la estética. Mediante la técnica quirúrgica Le Fort III se ha demostrado evidente mejoría de los parámetros respiratorios para tratar las apneas obstructivas durante el sueño7. En una segunda fase se puede tratar la hipoplasia maxilar mediante un avance cigomático-maxilar. Se consideran de interés los estudios campimétricos, capaces de evidenciar déficits que los estudios de visión central no detectan8. Requieren un seguimiento interdisciplinar, abarcando también factores psicológicos para facilitar su integración social9.
Es necesario el asesoramiento genético dado que el riesgo de recurrencia para el afectado es del 50% en cada gestación, sin poder predecir su gravedad. El estudio genético prenatal y/o preimplantacional solo puede realizarse cuando se conoce la mutación patogénica del caso índice. La ecografía prenatal de alta resolución puede detectar el exoftalmos o el hipertelorismo; sin embargo, la craneosinostosis solo es detectable mediante ecografía tridimensional o resonancia magnética (RM). En las formas de novo, al no poder descartarse un mosaicismo germinal, el riesgo de recurrencia para los padres en futuros embarazos se estima inferior al 3%, por lo que se aconseja realizar estudio prenatal10.
Del resto de entidades alélicas de los genes FGFR1, FGFR2 y FGFR3 que tienen en común la craneosinostosis, se resume a continuación lo más característico de cada síndrome, ordenándose, como en la tabla 1, en primer lugar los afectados por FGFR2 y otros posibles genes, y al final los afectados por FGFR3.
El síndrome de Apert (MIM 101200) es debido a mutaciones heterocigotas del gen FGFR2, de herencia AD, aunque predominan los casos debidos a mutación de novo. Lo más característico es la presencia de sindactilia severa en las manos y los pies. Son frecuentes las malposiciones dentales con dientes apiñados11.
El síndrome de Pfeiffer (MIM 101600) es debido a mutaciones del gen FGFR2 (Pfeiffer tipo 2) (MIM 176943) y más raramente del FGFR1 (8p12-p11.2) (Pfeiffer tipo 1) (MIM 136350). Descrito en 1964, se han publicado poco más de 60 casos. De herencia AD, con penetrancia y expresividad variable incluso intrafamiliar (se han descrito familias en las que un miembro padecía afectación digital sin craneosinostosis, y en otro miembro al revés o con mínima afectación). Se conoce la existencia de que una misma mutación en FGFR2 con fenotipo Pfeiffer, se haya descrito previamente con fenotipo Crouzon, Jackson-Weiss o Saethre-Chotzen12.
El síndrome de Jackson-Weiss (MIM 123150) se debe a mutaciones del gen FGFR2. Solo un paciente descrito con una mutación (P252R) en el gen FGFR1 (8p11.2-p11.1)13. De herencia AD, también con gran variabilidad de la expresividad intrafamiliar.
El síndrome de Saethre-Chotzen (también llamado cráneo-óculo-dental) (MIM 101400) se debe a mutaciones del gen TWIST1 (más de 35 conocidas y detectables en un 75%), y a mutaciones del gen FGFR2, habiéndose descrito solo 2 familias14,15. Aunque se ha estimado una incidencia entre 1:25.000 y 1:50.000 recién nacidos vivos, su frecuencia real se cree mayor por la existencia de pacientes sin diagnosticar con solo signos menores. Por otro lado, los avances genéticos han permitido constatar que en algunas casuísticas se incluía erróneamente a pacientes que padecían en realidad síndrome de Muenke, cuyo fenotipo puede ser similar16. La craneosinostosis suele requerir tratamiento quirúrgico precoz y casi la mitad de los pacientes precisan nuevas intervenciones antes de los 5 años al reaparecer la hipertensión intracraneal16.
El síndrome de Antley-Bixler se debe a mutaciones en el gen FGFR2, de herencia AD. Recientemente, se ha descrito genitales ambiguos en ambos sexos por deficiencia del citocromo P450 oxidoreductasa (POR), responsable de la síntesis de esteroides17,18. En estos nuevos casos se han podido encontrar mutaciones en el gen POR (7q11.2), de herencia AR (autosómica recesiva). Se han descrito mutaciones de este gen manifestando solo insuficiencia adrenal o infertilidad, sin dismorfias, y menor capacidad para metabolizar los fármacos que utilizan dicha vía metabólica19.
El síndrome de Beare-Stevenson es debido a un defecto del gen FGFR2 (se conocen 3 mutaciones de novo), o también del FGFR3. Solo se han descrito 12 casos esporádicos desde la primera descripción en 1969. Son características las anomalías cutáneas como cutis gyrata (arrugas y piel sobrante) en cráneo, cara y extremidades, y acantosis nigricans. El primer paciente descrito por Beare en 1969 y otros 2 casos20 presentaban dientes neonatales con posterior hipodontia.
El síndrome de Crouzon con acantosis nigricans es debido a la mutación recurrente G1172A, missense específica, en el dominio transmembrana del gen FGFR3. Publicados hasta la fecha 35 casos. Tienen fenotipo Crouzon de diversa gravedad al que se le añade una acantosis nigricans aparecida durante los primeros 2-3 años de vida (como en nuestro segundo paciente), aunque se conoce un caso de presentación neonatal21. El 5% de los pacientes con síndrome de Crouzon presentan acantosis nigricans4. Se puede localizar en la región posterior del cuello, espalda, axilas, ingles, perioral, periorbitaria, pliegues nasolabiales, tórax o abdomen. Puede evolucionar en años a una hiperplasia verrucosa. Se ha descartado una insensibilidad a la insulina, siendo la hipótesis más probable una disfunción de melanocitos, raramente cementomas mandibulares o desmoosteoblastomas5. Tiene un amplio espectro clínico por probable existencia de genes modificadores22,23. Se ha descrito otra mutación en el mismo gen, también de herencia AD, en la que solo se expresa la anomalía cutánea.
El síndrome de Muenke también es debido a una mutación recurrente (P250R) del gen FGFR324, de herencia AD, con baja penetrancia y expresividad muy variable. Un 61% son casos esporádicos. Se han publicado más de 300 casos. Es muy frecuente la sordera, especialmente para bajas frecuencias, y retraso psicomotor en la mitad de los pacientes. Posible dismorfismo sexual en varones. Una mutación diferente en el mismo gen, la P250L, da lugar a un fenotipo muy similar a Muenke. Se han descrito casos en los que el único síntoma clínico es la sordera. Es muy importante la confirmación genética, especialmente en los casos con poca expresividad clínica, permitiendo realizar un correcto asesoramiento genético25.
Existe aún otra entidad denominada osteoglophonic dwarfism (o enanismo en «sacabocados») (8p11.1-p11.2) (MIM 66250), con fenotipo de acondroplasia pero asociando también craneosinostosis y lesiones quísticas metafisarias aparecidas generalmente en la etapa adulta. Al resultar el nanismo el signo guía principal, no la hemos incluido en la tabla, pero también constituye otra craneosinostosis alélica de cualquiera de los genes FGFR1, FGFR2 o FGFR3.
En conclusión, las craneosinostosis pueden presentarse aisladas o formar parte de un síndrome complejo. En el primer grupo, un 10-14% también son de causa genética. En las formas sindrómicas es necesario detallar los signos clínicos pues en ocasiones pueden orientar el diagnóstico. Sin embargo, dada la frecuente existencia de signos clínicos comunes entre algunas de las entidades, es recomendable realizar el estudio molecular para intentar confirmarlo, lo que permitirá ofrecer a las familias un correcto asesoramiento genético9. A partir de los 2 pacientes presentados con fenotipo Crouzon, se han esquematizado los síndromes craneoestenóticos que son formas alélicas de los genes FGFR1, FGFR2 y FGFR3, resumiéndose en la tabla 1 los principales aspectos clínicos, genes implicados y porcentaje de detección de mutaciones2, siendo también llamativa su amplia heterogeneidad genética y alélica. Serán necesarias más investigaciones para profundizar en la fisiopatología de estas entidades e intentar mejorar la relación fenotipo-genotipo.
Conflictos de interesesLos autores declaran no tener ningún conflicto de intereses.
