



La sarcoidosis es una enfermedad multisistémica rara, caracterizada por la formación de granulomas no caseificantes en diferentes tejidos1,2. Existen 2formas, la sarcoidosis de inicio precoz o síndrome de Blau, y la forma juvenil, de causa desconocida y heterogénea clínicamente, aunque las manifestaciones pulmonares3,4 son las más frecuentes. La afectación esplénica es rara, habiéndose publicado casos aislados3.
Presentamos 2casos pediátricos de sarcoidosis que iniciaron con esplenomegalia masiva.
Caso 1: niño de 11 años, sin antecedentes de interés, que presentaba fiebre de 12 días de evolución, adenopatías pequeñas submandibulares, axilares e inguinales y hepatoesplenomegalia palpable hasta la fosa ilíaca. Analíticamente, destacaban leucocitopenia (1.570/mm3), neutropenia (610/mm3) y trombocitopenia (77.000/mm3), confirmadas en frotis de sangre periférica, así como creatinina de 0,93mg/dl e hipertransaminasemia (GOT 73 U/l, GPT 85 U/l). No presentaba elevación de PCR/VSG. La radiografía de tórax fue anodina y la ecografía abdominal confirmó hepatoesplenomegalia.
Serologías, cultivos y PCR virales, incluyendo CMV y VEB, fueron negativos; así como Mantoux y QuantiFERON. Los valores de las inmunoglobulinas fueron normales. Presentaba una cifra de enzima conversora de angiotensina (ECA) en 248,9 U/l (valores normales 8-55 U/l) e hipercalcemia (10,86mg/dl).
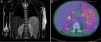
Se realizó una resonancia magnética toraco-abdominal y una tomografía por emisión de positrones (fig. 1).
En la biopsia de médula ósea se observaron microgranulomas y ausencia de células neoplásicas; en la biopsia ganglionar, cuerpos de Schaumann.
Ante estos resultados, tras descartar etiología infecciosa y tumoral se llegó al diagnóstico de sarcoidosis.
Durante el diagnóstico, espontáneamente, desaparecieron la fiebre y las alteraciones analíticas, disminuyó la hepatoesplenomegalia, interpretándose como un brote autolimitado.
A los 18 meses, reaparecieron leucocitopenia, hipertransaminasemia y elevación de ECA. Ecográficamente, presentaba nefrocalcinosis, iniciándose corticoides orales a 0,5mg/kg/día con buena evolución, aunque precisando metrotexato subcutáneo a 15mg/m2/semana con posterior cambio de este último a adalimumab por hipertransaminasemia. Durante su evolución presentó nuevo brote desencadenado por cuadro infeccioso, que evolucionó bien tras pauta de corticoides.
Caso 2: niño de 4 años en el que, 8 meses tras el diagnóstico de primoinfección por VEB confirmada serológicamente, persistió la hepatoesplenomegalia. En la ecografía abdominal presentaba esplenomegalia gigante y adenopatías mesentéricas, y mantenía 100.000 copias de VEB/ml en sangre, siendo el resto del cribado infeccioso negativo.
Al mes comenzó con fiebre y aumento de número y tamaño de adenopatías abdominales. La carga viral iba en descenso. Se realizó cribado de síndrome linfoproliferativo autoinmune (ALPS) y ligado al X, así como biopsia de médula y ganglionar para descartar malignidad, observándose granulomas epitelioides necrosantes.
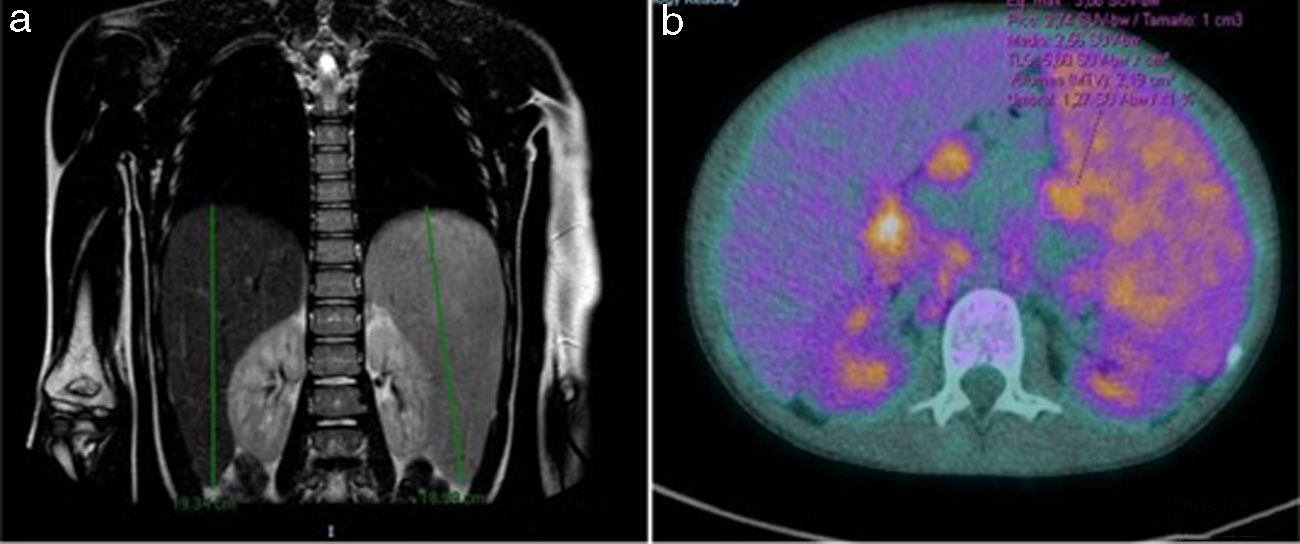
Tres meses después, siendo indetectable la carga de VEB en sangre, reinició fiebre, anemia e hipertransaminasemia. La tomografía computarizada toraco-abdominal mostró adenopatías mediastínicas y abdominales junto con 24 nódulos pulmonares (fig. 2a), realizándose nueva biopsia (adenopatía y bazo) que mostró adenitis granulomatosa inespecífica tipo sarcoidosis-like. Tanto Mantoux como QuantiFERON fueron negativos. Se realizó también LBA, que mostró una relación CD4/CD8 de 3,92 (valores normales 0,9-1,9). ECA en 239 U/l.
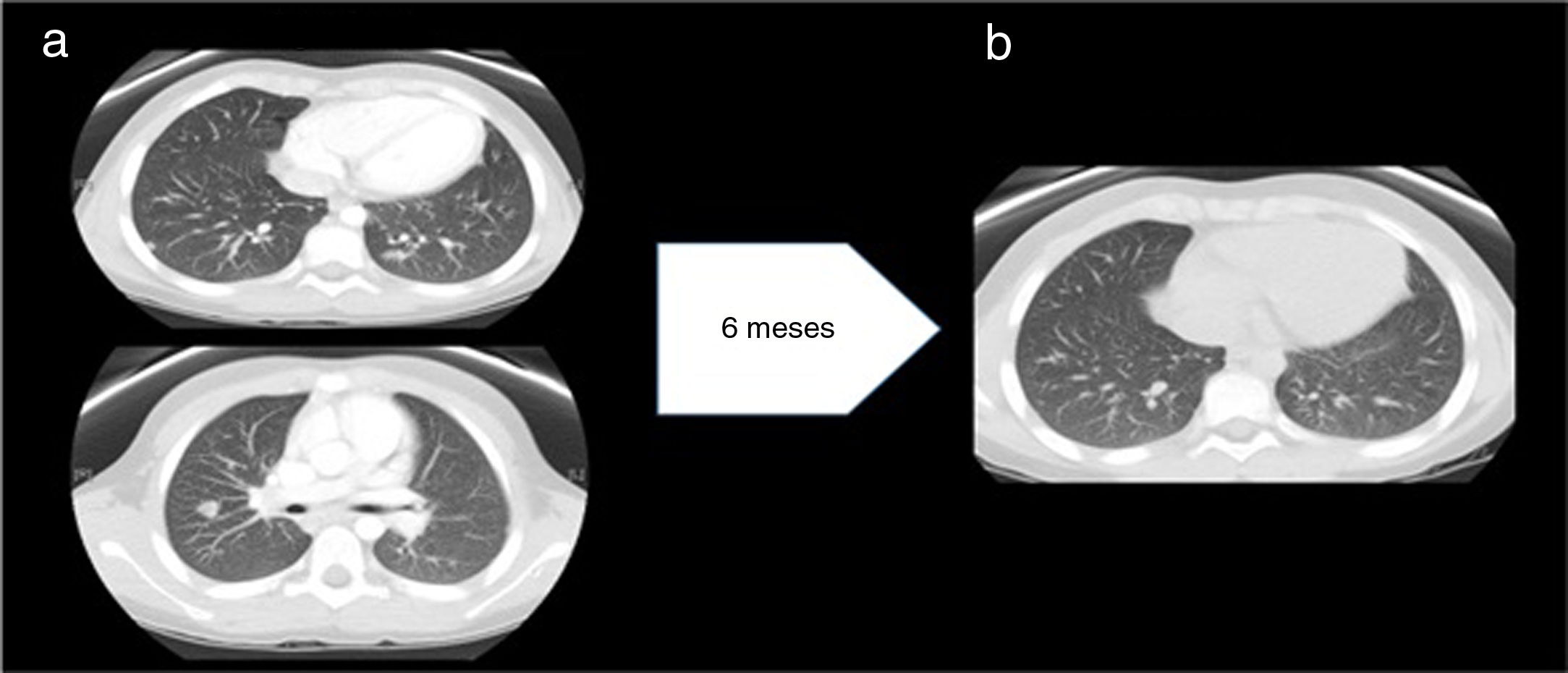
a) Antes del tratamiento con anti-TNF-α: múltiples nódulos, bien definidos, de localización subpleural, periféricos y parenquimatosos con tamaño máximo 11 mm. No se observan signos de fibrosis pulmonar. Hallazgos compatibles con sarcoidosis pulmonar estadio 2. b) Después del tratamiento con anti-TNF-α: disminución significativa en el número y tamaño de los nódulos pulmonares.
Con diagnóstico de sarcoidosis se iniciaron corticoides sistémicos (1mg/kg/día) y posteriormente metrotexato, disminuyendo el número de nódulos pulmonares y la esplenomegalia.
Aunque inicialmente el nivel de inmunoglobulinas fue normal, con la evolución desarrolló hipogammaglobulinemia. Tras intensificar tratamiento inmunosupresor con adalimumab y administrar varias dosis de inmunoglobulinas por vía intravenosa, recuperó niveles, manteniéndose a lo largo del tiempo sin inmunoglobulina sustitutiva, descartándose IDCV. Quedó asintomático y disminuyó de forma significativa el número de nódulos tras 10 años de seguimiento (fig. 2b).
En conclusión, la sarcoidosis es una enfermedad infrecuente en pediatría que presenta una gran heterogeneidad clínica1,2. La esplenomegalia, descrita en adultos en menos del 10%, puede aparecer hasta en el 50% en niños, a veces de gran tamaño, como ocurrió en nuestros pacientes. Esto obliga a realizar un amplio diagnóstico diferencial1 con la etiología tumoral y ALPS, especialmente si hay citopenias, y con infecciones (especialmente tuberculosis) e inmunodeficiencias cuando hay afectación pulmonar. La IDCV tiene una presentación muy similar5, pudiendo iniciar como esplenomegalia, y siendo la enfermedad pulmonar intersticial linfocítica-granulomatosa una de sus principales complicaciones, por lo que es fundamental realizar niveles de inmunoglobulinas antes de iniciar terapia inmunosupresora. Por otra parte, el síndrome linfoproliferativo ligado al X debe considerarse en varones si el desencadenante es el VEB, como ocurrió en el segundo caso.
La nefrocalcinosis es una complicación descrita hasta en un 10% de los casos4, de ahí la necesidad de seguimiento para orientar decisiones terapéuticas y evitar secuelas.
En cuanto al tratamiento, no se encuentra protocolizado en pediatría. Al igual que en adultos, inicialmente se basa en corticoterapia3,6. En casos refractarios o corticodependientes, se valora la posibilidad de asociar inmunosupresores como metotrexato o anti-TNF-α6.
Presentaciones previas: 46.a Reunión anual SPAO. Jaén, marzo del 2019. 67.° Congreso de la Asociación Española de Pediatría. Burgos, junio del 2019.
