



Sarcoidosis is a rare multisystemic disease characterized by the development of noncaseating granulomas in different tissues.1,2 There are 2 forms of juvenile sarcoidosis: early-onset sarcoidosis or Blau syndrome, and late-onset sarcoidosis, which is of unknown aetiology and has a heterogeneous clinical presentation, although pulmonary manifestations are most frequent.3,4 Splenic involvement is rare, and only a few cases of it have been published in the literature.3
We present 2 cases of juvenile sarcoidosis with onset with massive splenomegaly.
Case 1: boy aged 11 years with no relevant history that presented with fever of 12 days’ duration, mild lymph node enlargement in the submandibular, axillary and inguinal regions, and hepatosplenomegaly detectable on palpation through the iliac fossa. The salient findings of blood tests were leukopenia (1570/mm3), neutropenia (610/mm3) and thrombocytopenia (77000/mm3), confirmed by examination of a peripheral blood smear, a creatinine level of 0.93mg/dL and hypertransaminasaemia (aspartame aminotransferase, 73U/L; alanine aminotransferase, 85U/L). There was no elevation of C-reactive protein or the erythrocyte sedimentation rate. The findings of the chest radiograph were unremarkable, and the abdominal ultrasound scan confirmed the presence of hepatosplenomegaly.
Serological tests, cultures and viral polymerase chain reaction tests for viral detection (including cytomegalovirus and Epstein–Barr virus [EBV]), as were the Mantoux test and QuantiFERON assay. Immunoglobulin levels were normal. The patient had an elevated level of angiotensin converting enzyme (ACE, 248.9U/L; normal range, 8–55U/L) and hypercalcaemia (10.86mg/dL).
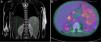
The patient underwent a magnetic resonance scan of the thorax and abdomen and a positron emission tomography (PET) scan (Fig. 1).
![(a) Late-onset hepatosplenomegaly. No signs of pulmonary involvement. (b) Hypermetabolic lymphadenopathy (maximum standardized uptake value [SUVmax], 3.89). Hypermetabolic splenomegaly (SUVmax, 3.15).](https://static.elsevier.es/multimedia/23412879/0000009400000001/v1_202101160808/S234128792030209X/v1_202101160808/en/main.assets/gr1.jpeg?xkr=ue/ImdikoIMrsJoerZ+w9/qVHBXBqbSQ7FNUvNof+6+l4v03CmyaR9Rm+q8TRfDERovN5wMUmDVyYvA+rI0XvO+/Wy1mIFlmfA3Y/3zTyE8akmTxw9WZJmePo44donQ0zjakTiapnMn+DCUTNZCMjKq7vAAjVSKCuTdr2bVDQW8CnBKlzeVabTh3KpzvbZo5RQs0Yv+snm0aR4KOdgj4MH/J7ohfmM+beYZxtVvvron0SpMsLQPnXfLF6vpELSY148G8p9lmtyFLClEA8Jioh8kA5JV/epGg17SDaiEmgSo=)
The bone marrow biopsy revealed an absence of cancerous cells, and the lymph node biopsy the presence of Schaumann bodies.
These findings, having ruled out an infectious or cancerous aetiology, led to diagnosis of sarcoidosis.
During the diagnostic process, the fever and blood test abnormalities resolved, and the hepatosplenomegaly improved, so the episode was considered to be self-limited.
At 18 months, the patient experienced a recurrence of leukopenia, hypertransaminasaemia and elevation of ACE. The ultrasound scan revealed nephrocalcinosis. This led to initiation of treatment with oral corticosteroids at a dose of 0.5mg/kg/day, with a positive response, although the patient also required methotrexate delivered subcutaneously at a dose of 15mg/m2/week that was eventually switched to adalimumab due to the presence of hypertransaminasaemia. During the follow-up, the patient developed a new episode triggered by infection that resolved with corticosteroids.
Case 2: boy aged 4 years with persisting hepatosplenomegaly 8 months after diagnosis of primary infection by EBV with serological confirmation. The abdominal ultrasound scan revealed massive splenomegaly and mesenteric lymphadenopathy, and the EBV load in blood remained detectable at 100000copies/mL, while all other infection screening tests were negative.
A month later, the patient developed fever and the number and size of involved lymph nodes in the abdominal region increased. The blood EBV load exhibited a decreasing trend. The patient underwent screening for X-linked autoimmune lymphoproliferative syndrome (ALPS) and bone marrow and lymph node biopsies, with detection of necrotising epithelioid granulomas.
Three months later, when the EBV load in blood was undetectable, the patient experienced a recurrence of fever accompanied by anemia and hypertransaminasaemia. The computed tomography scan of the thorax and abdomen revealed mediastinal and abdominal lymphadenopathy and 24 nodules in the lungs (Fig. 2a), which led to performance of additional biopsies (lymph node and spleen) that revealed sarcoid-like nonspecific granulomatous lymphadenitis. The Mantoux test and QuantiFERON assay were both negative. Examination of a bronchoalveolar lavage sample revealed a CD4/CD8 ratio of 3.92 (normal range, 0.9–1.9). The serum level of ACE was 239U/L.
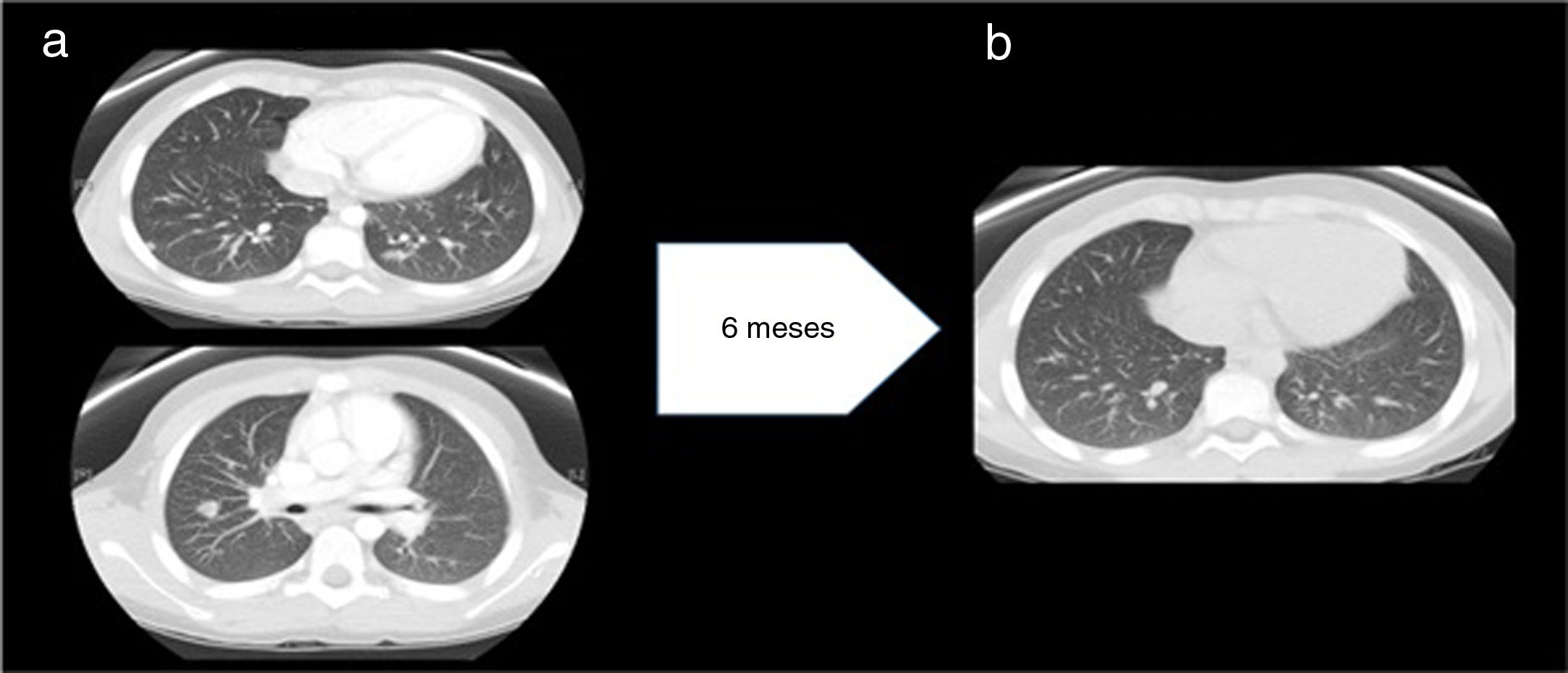
(a) Before treatment with anti-TNF-α: multiple well-delimited parenchymal nodules with a subpleural, peripheral location and a maximum diameter of 11mm. No evidence of pulmonary fibrosis. Findings compatible with stage 2 pulmonary sarcoidosis. (b) After treatment with anti-TNF-α: significant decrease in the number and size of pulmonary nodules.
Following diagnosis of sarcoidosis, the patient started treatment with systemic corticosteroids (1mg/kg/day) and later on with methotrexate, which achieved a reduction in the number of pulmonary nodules and in the size of the spleen.
Although in the initial evaluation the level of immunoglobulins had been within the normal range, during the follow-up the patient developed hypogammaglobulinemia. After intensification of immunosuppression with adalimumab and administration of several doses of intravenous immunoglobulin, the levels recovered and remained normal over time without immunoglobulin replacement therapy, which ruled out common variable immune deficiency. The patient became asymptomatic and the number of nodules had decreased significantly at 10 years of follow-up (Fig. 2b).
In conclusion, sarcoidosis is a disease that is rare and exhibits a heterogeneous clinical presentation in the pediatric age group.1,2 Splenomegaly, described in fewer than 10% of affected adults, may be present in up to 50% of pediatric cases, and be severe in some, as was the case in our sample. This entails an extensive differential diagnosis1 including neoplasias and ALPS, especially in the presence of cytopenia, and infection (especially tuberculosis) and immune deficiencies in case of pulmonary involvement. Common variable immune deficiency has a similar presentation,5 as it may have onset with splenomegaly and one of its most frequent complications is granulomatous-lymphocytic interstitial lung disease, so it is essential that immunoglobulin levels are measured before initiating immunosuppressive therapy. On the other hand, X-linked lymphoproliferative disease should be considered in male patients if the trigger is EBV infection, as was the case of the second patient presented here.
Nephrocalcinosis is a complication described in up to 10% of cases,4 highlighting the need of monitoring to guide treatment planning and prevent sequelae.
As for treatment, there is no standardized approach to the management of systemic sarcoidosis in the pediatric population. Steroid therapy is the first-line treatment, as is the case in adult patients.3,6 In refractory or steroid-dependent cases, there is the option of adding an immunosuppressive agent such as methotrexate or anti-TNF-α.6
Please cite this article as: Cobreros-Pérez Á, Galindo-Zavala R, Carazo-Gallego B, Martín-Pedraz L, Núñez-Cuadros E. Sarcoidosis sistémica: cuando la esplenomegalia no es lo que parece. An Pediatr (Barc). 2021;94:48–50.
Previous presentations: this study was presented at the 46th Annual Meeting of the Sociedad de Pediatría de Andalucía Oriental, March 2019, Jaen, Spain; and the 67th Congress of the Asociación Española de Pediatría, June 2019, Burgos, Spain.

Anales de Pediatría (English Edition) follows the Recommendations for the Conduct, Reporting, Editing and Publication of Scholarly Work in Medical Journals