



El diagnóstico de fibrosis quística (FQ) a través del cribado neonatal (CN) está bien establecido en muchos países y brinda la oportunidad de un diagnóstico y tratamiento temprano antes del desarrollo de daño estructural pulmonar irreversible.
En 1999, Cataluña, Castilla-León y las Islas Baleares iniciaron el programa CN para FQ. En los últimos 10 años su implementación se extendió rápidamente y todas las autonomías ofrecen el programa CN para FQ desde 2015. Hay varias estrategias diferentes en toda España. Creemos que es muy oportuno contar con una guía actualizada y consensuada para el diagnóstico, el seguimiento y el tratamiento de los pacientes diagnosticados de FQ mediante CN.
Newborn screening (NBS) for cystic fibrosis (CF) is well-established in many countries and provides the opportunity for an early diagnosis and treatment before the development of irreversible structural lung damage.
In 1999, Catalonia, Castilla-León, and the Balearic Islands started the NBS programme for CF. In the last 10 years its implementation rapidly spread and all the autonomies offer the NBS programme for CF since 2015. There are many different strategies across Spain. It is believed that it is very opportune to have an updated and consensual guide for the diagnosis, follow-up, and treatment of patients diagnosed by neonatal screening.
La fibrosis quística (FQ) es la enfermedad genética grave con patrón de herencia autosómica recesiva más frecuente en la población caucásica, con una incidencia de 1 de cada 1.800-25.000 recién nacidos (RN) dependiendo de la región o etnia de origen. En el año 2016 la Canadian Cystic Fibrosis Foundation publicó una mediana de supervivencia de 53,3 años1. El aumento tan importante de la supervivencia de estos pacientes es debido a una serie de factores a los que ha contribuido de forma determinante la implementación del diagnóstico precoz a través del cribado neonatal (CN) del RN.
La FQ cumple los requisitos para que su detección precoz y el CN está justificado para conocer la incidencia real de la enfermedad, para un asesoramiento genético precoz y para iniciar un tratamiento inmediato destinado a prevenir o minimizar el daño pulmonar, ya que en la actualidad hay disponibles moléculas dirigidas a corregir el defecto en la proteína CFTR defectuosa.
Desde el año 2015 el CN se lleva a cabo en todas las comunidades autónomas de España.
Creemos que es muy oportuno disponer de una guía actualizada y consensuada para el diagnóstico, seguimiento y tratamiento de aquellos casos que cumplen los requisitos diagnósticos. El trabajo describe de forma resumida todos los aspectos multidisciplinares para el manejo de estos pacientes.
Beneficios y riesgos del cribado neonatal de fibrosis quísticaEn la actualidad, la mayoría de los profesionales dedicados al cuidado de los pacientes con FQ están de acuerdo en que un programa de CN bien diseñado conlleva un balance beneficio/riesgo positivo en el que los beneficios (nutricionales, respiratorios, de erradicación precoz de microorganismos, asesoramiento genético y participación en ensayos clínicos de intervención precoz) superan a las desventajas (sobre todo en el ámbito psicosocial), y que resulta coste-efectivo a largo plazo2-5. Para garantizar los beneficios de un CN, una vez hecho el diagnóstico, los pacientes con FQ deben ser controlados adecuadamente (siguiendo los estándares estatales de cuidado) en unidades de referencia de FQ6,7. La mayoría de los riesgos del CN se minimizan si se llevan a cabo las siguientes actuaciones6: a) instaurar un programa de CN adaptado a las características poblacionales, b) ofrecer comunicación e información efectiva a los padres durante el proceso diagnóstico, tanto en afectos como en portadores o falsos positivos, intentando disminuir el tiempo de espera para el diagnóstico definitivo al mínimo posible; c) ofertar un seguimiento clínico en estos niños.
Protocolos de cribado neonatal en EspañaTripsina (tripsinógeno) inmunorreactivaLos niveles en suero de la tripsina inmunorreactiva (TIR) son más altos en los RN con FQ y permanecen elevados más tiempo que en aquellos que no están afectados por la enfermedad8,9.
Un nivel elevado de TIR al nacimiento no es específico de FQ, ya que hay RN sanos que muestran elevaciones transitorias de esta enzima. La especificidad de una única muestra elevada de TIR es baja, por lo que se han desarrollado protocolos basados en el análisis del ADN en la primera muestra del RN (TIR+ADN) que es el más implementado en España, o en la solicitud de una segunda muestra para medir la TIR con el análisis posterior del ADN (TIR+TIR+ADN), o la prueba del sudor (fig. 1). Cabe recordar que en los lactantes con íleo meconial los valores de TIR son normales en hasta un 30% de los casos. La estrategia TIR+TIR solo se emplea en algunas comunidades autónomas.

Protocolos de actuación en el diagnóstico por cribado neonatal de la fibrosis quística propuestos en la European best practice guidelines for cystic fibrosis neonatal screening7 y que se realizan en España.
En España existen 3 estrategias: TIR+TIR, TIR+TIR+ADN y TIR+ADN+TIR.
Estudio genético de fibrosis quísticaEl estudio genético forma parte de la mayoría de los programas de CN de FQ y se realiza en aquellas muestras que presentan un nivel de TIR superior al punto de corte decidido. Cada población deberá aplicar un programa de acuerdo con su espectro mutacional.
Tras el estudio genético a un RN con una TIR elevada, son posibles 4resultados:
- a)
Si se encuentran 2 mutaciones causantes de FQ, se debe realizar la prueba del sudor y el estudio de segregación, a fin de corroborar que cada una de las mutaciones procede de uno de sus progenitores.
- b)
Si se encuentra una sola mutación, debe realizarse la prueba del sudor. Si la prueba del sudor es dudosa, se debe ampliar el estudio molecular para intentar caracterizar la segunda mutación. Si la prueba del sudor es negativa, se asume al RN como portador.
- c)
Si no se encuentran mutaciones y la prueba del sudor es normal, estos serían los falsos positivos del programa.
Otro grupo es el de aquellos neonatos en los que no se llega a alcanzar un diagnóstico concluyente, ni siquiera mediante pruebas confirmatorias, y para los que recientemente el grupo de trabajo europeo de CN de FQ ha propuesto el término CFSPID (del inglés cystic fibrosis screen positive, inconclusive diagnosis)10. Este grupo incluye:
- a)
RN en los que se ha encontrado una mutación CFTR en solo un alelo y valores intermedios de cloro en sudor (30-59 mmol/L).
- b)
Aquellos que presentan una mutación CFTR en cada alelo, pero solo una de ellas está clasificada como causante de FQ, y valores de cloro en sudor normales (<30 mmol/L).
Se debe hacer un seguimiento de este último grupo de pacientes de forma regular, ya que en algunos casos se realiza el diagnóstico de FQ a edades posteriores por aparición de síntomas y elevación del cloro en sudor.
Prueba del sudorLa prueba del sudor es la piedra angular del diagnóstico de la FQ. Los RN con una prueba de CN de FQ positiva deben ser remitidos para una prueba del sudor únicamente a unidades de FQ con una experiencia acreditada en el diagnóstico.
La prueba del sudor tiene 3fases: 1) Estimulación mediante iontoforesis con pilocarpina. 2) Recogida de la muestra durante 30 min mediante uno de 2métodos: el sistema Macroduct o bien papel de filtro o prepesados (sistema original de Gibson y Cooke). El volumen de la muestra debe ser de, al menos, 15μL (Macroduct) o 75 mg (gasa). 3) Determinación de la concentración de cloro mediante titulación por coulometría en cloridómetro11.
No se acepta para la confirmación del diagnóstico determinar únicamente la conductividad del sudor mediante nanoduct o sweat check (Macroduct). Las muestras deben ser analizadas inmediatamente.
La prueba se clasifica como «normal» si el valor de cloro es < 30 mmol/L; «intermedia» si es 30-59 mmol/L y «positiva», si es ≥60 mmol/L12.
Aquellos en los que persisten valores intermedios deben ser estudiados, incluyendo un estudio genético ampliado y la repetición de la prueba del sudor a los 6 meses.
Visita informativa sobre el resultado del cribado neonatalLa realización del CN suscita un grado de ansiedad en los padres. Por ello es recomendable que el resultado se comunique pronto, a ser posible el mismo día en que se complete el estudio.
Si el RN está afecto de FQ, se dará una información general de todas las pruebas realizadas de la enfermedad, así como de los resultados esperanzadores de los nuevos tratamientos y de la extensa investigación que se lleva a cabo, lo que proporciona una nueva perspectiva optimista sobre la enfermedad.
Al final de la visita, si el RN no está afecto, se emitirá un informe en el que se refleje que el niño no tiene FQ o que es portador y se ofrecerá asesoramiento genético a los padres13.
Visita inicial del recién nacido con fibrosis quísticaEl diagnóstico de FQ mediante CN es inesperado para los padres, ya que, en la mayoría de las ocasiones, el RN todavía no presenta manifestaciones clínicas. Por ello, debe manejarse cuidadosamente el impacto psicosocial que produce en la familia14.
Los aspectos más importantes que tratar son:
- •
Dar una visión general de la enfermedad.
- •
Introducir el concepto de «equipo de cuidados».
- •
Manejar las expectativas de la familia.
- •
Ofrecer apoyo psicológico.
Uno de los aspectos destacados de la visita es que la información aportada sea básica, positiva, sensible y empática, expuesta con términos fácilmente comprensibles15.
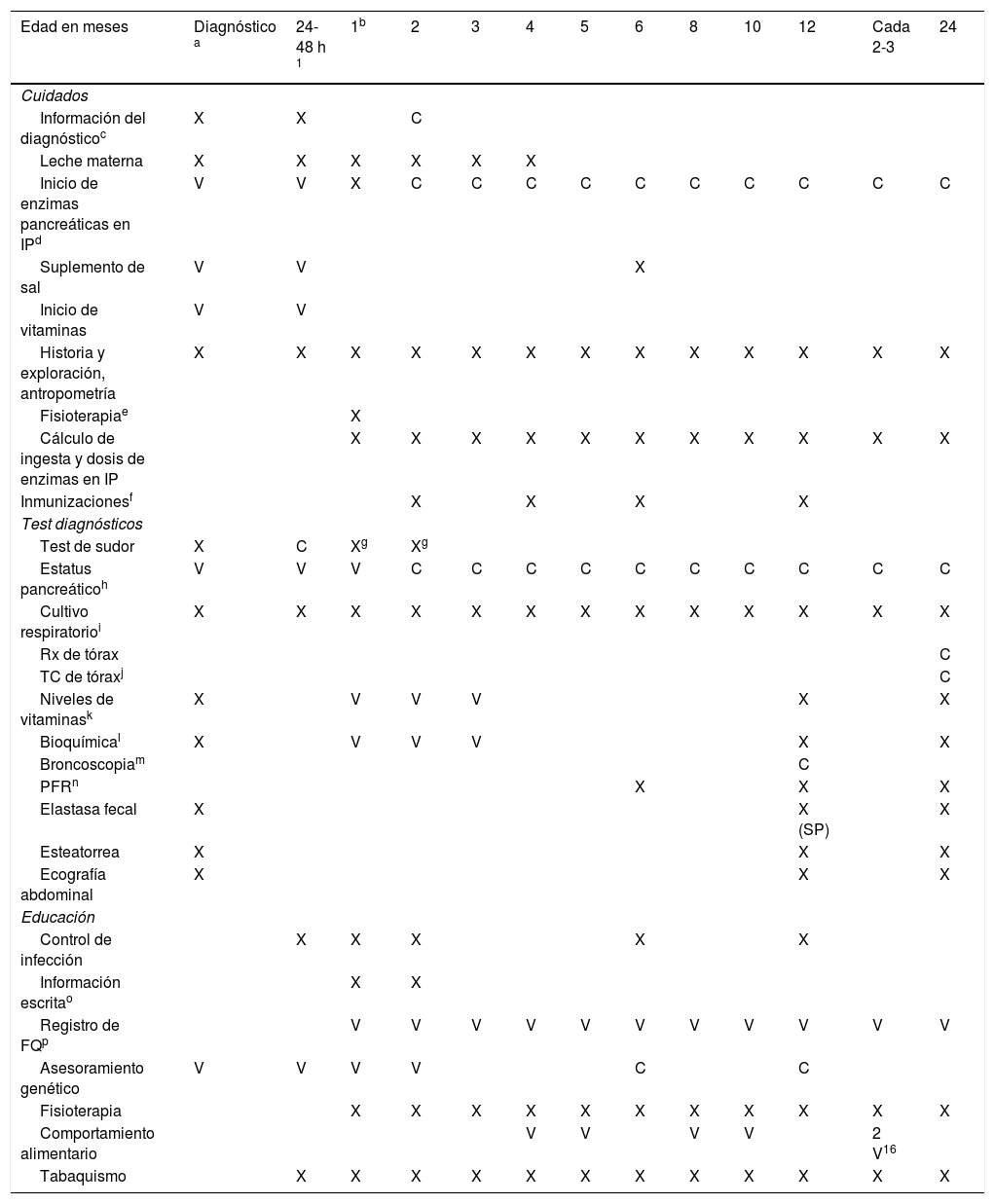
En la tabla 1 se especifican los procedimientos en la primera visita.
Calendario de visitas programadas
| Edad en meses | Diagnóstico a | 24-48 h 1 | 1b | 2 | 3 | 4 | 5 | 6 | 8 | 10 | 12 | Cada 2-3 | 24 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cuidados | |||||||||||||
| Información del diagnósticoc | X | X | C | ||||||||||
| Leche materna | X | X | X | X | X | X | |||||||
| Inicio de enzimas pancreáticas en IPd | V | V | X | C | C | C | C | C | C | C | C | C | C |
| Suplemento de sal | V | V | X | ||||||||||
| Inicio de vitaminas | V | V | |||||||||||
| Historia y exploración, antropometría | X | X | X | X | X | X | X | X | X | X | X | X | X |
| Fisioterapiae | X | ||||||||||||
| Cálculo de ingesta y dosis de enzimas en IP | X | X | X | X | X | X | X | X | X | X | X | ||
| Inmunizacionesf | X | X | X | X | |||||||||
| Test diagnósticos | |||||||||||||
| Test de sudor | X | C | Xg | Xg | |||||||||
| Estatus pancreáticoh | V | V | V | C | C | C | C | C | C | C | C | C | C |
| Cultivo respiratorioi | X | X | X | X | X | X | X | X | X | X | X | X | X |
| Rx de tórax | C | ||||||||||||
| TC de tóraxj | C | ||||||||||||
| Niveles de vitaminask | X | V | V | V | X | X | |||||||
| Bioquímical | X | V | V | V | X | X | |||||||
| Broncoscopiam | C | ||||||||||||
| PFRn | X | X | X | ||||||||||
| Elastasa fecal | X | X (SP) | X | ||||||||||
| Esteatorrea | X | X | X | ||||||||||
| Ecografía abdominal | X | X | X | ||||||||||
| Educación | |||||||||||||
| Control de infección | X | X | X | X | X | ||||||||
| Información escritao | X | X | |||||||||||
| Registro de FQp | V | V | V | V | V | V | V | V | V | V | V | ||
| Asesoramiento genético | V | V | V | V | C | C | |||||||
| Fisioterapia | X | X | X | X | X | X | X | X | X | X | X | ||
| Comportamiento alimentario | V | V | V | V | 2 V16 | ||||||||
| Tabaquismo | X | X | X | X | X | X | X | X | X | X | X | X |
C: considerar realizarlo en esta visita; V: realizar en una de estas visitas. X: realizar en esta visita.
Explicar por qué sabemos que su hijo tiene FQ y dar detalles del diagnóstico, genética y repercusión en otros hermanos y familiares.
Iniciar enzimas pancreáticas si el paciente tiene síntomas de malabsorción o elastasa fecal < 200 mcg/g o 2 mutaciones CFTR asociadas a IP.
Instrucción en técnicas que facilitan el aclaramiento: percusión y drenaje postural. Existe controversia acerca de la más apropiada en niños asintomáticos.
Coordinación con visitas habituales en atención primaria. Incluir varicela, S. pneumoniae y rotavirus. Gripe en mayores de 6 meses y palivizumab en menores de un año.
Si la IP no está presente al diagnóstico, repetir elastasa fecal al menos 2veces en el primer año de vida y anualmente tras el primer año, ya que la IP puede aparecer después.
Considerar realizar con mayor frecuencia el cultivo de muestras respiratorias si el lactante presenta síntomas.
Aconsejable de manera rutinaria a partir de los 2 años si el niño está asintomático, o antes, si hay sintomatología. TC espiratoria, bajas dosis de radiación y cortes espiratorios para valorar la enfermedad de las vías aéreas periféricas.
Repetir a los 1-2 meses del inicio de la suplementación. Medir con mayor frecuencia si los niveles son anormales.
Hemograma, electrólitos, urea, creatinina, albúmina, AST, ALT, GGT, bilirrubina, fosfatasa alcalina (FA).
Algunos centros practican lavado broncoalveolar alrededor del tercer mes y al año de vida para detección precoz de infección, de marcadores de inflamación o de aspiración/reflujo, así como biopsia bronquial con fines de investigación clínica.
Pruebas de función respiratoria en lactantes: considerar hacerlas en los centros con disponibilidad.
Entregar folleto con indicaciones de cómo y cuándo debe contactarse con la unidad de FQ (diario de síntomas y tratamientos prescritos y recibidos fuera de las citas programadas).
Obtención de consentimiento informado para el registro de FQ.
Fuente: Adaptado de Borowitz et al.14.
Se recomienda un seguimiento clínico por el gastroenterólogo pediátrico cada 1-2 semanas tras el diagnóstico a fin de asegurar una nutrición adecuada y, luego, mensual en los 3 primeros meses. Si el estado del paciente es el adecuado, los controles posteriores pueden realizarse cada 2-3 meses.
NutriciónEl principal objetivo de la intervención nutricional en la FQ consiste en promover el crecimiento y el desarrollo normales para la edad (tabla 2).
Guía para indicación de intervención nutricional18
| <2 años | 2-18 años | |
|---|---|---|
| Estado nutricional óptimo: consejo nutricional preventivo | Peso para talla ≥ P50 | IMC ≥ P50 |
| Estado nutricional subóptimo: modificaciones dietéticas y suplemento nutricional oral | Peso para talla en P10-P50 Pérdida de peso Fallo de medro | IMC P10-P50 Pérdida de peso en 2-4 meses o ausencia de ganancia en 2 meses |
| Malnutrición persistente: nutrición enteral (sonda nasogástrica o gastrostomía) | Peso para talla < P10 Pérdida de peso persistente o fallo de medro a pesar de suplemento oral | IMC < P10 Caída de 2 percentiles a pesar de suplemento oral |
IMC: índice de masa corporal, calculado como peso (kg)/talla (m2).
Se recomienda fomentar la lactancia materna a demanda. Independientemente del tipo de lactancia, se administrarán enzimas pancreáticas en los pacientes con insuficiencia pancreática (IP)16,17.
La introducción de la alimentación complementaria debe hacerse de forma habitual a partir de los 6 meses en caso de lactancia materna exclusiva y entre los 4 y los 6 meses en los alimentados con fórmula artificial18,19.
Función pancreática y hepáticaYa en el periodo neonatal, los niños afectos de FQ pueden presentar hipovitaminosis e IP.
En el caso de advertir clínicamente una probable IP, o tener 2mutaciones que se asocien a ella, se iniciará la administración de enzimas pancreáticas y vitaminas (tabla 3) aunque, si bien no se debe retrasar, lo preferible y aconsejable es tener el resultado de la elastasa fecal para iniciar el tratamiento.
Recomendaciones de administración de terapia de sustitución pancreática en niños con insuficiencia pancreática
| Niños de 0-12 meses | 2.000-4.000 UI lipasa/120 ml o toma de lactancia materna |
| Niños de 12 meses a 4 años | 1.000 UI lipasa/kg por tomaa, máximo 2.500 UI/kg al día o 10.000 UI/kg al día o 4.000 UI/g grasa dieta |
| Niños >4 años a adultos | 500 UI lipasa/kg por tomaa, máximo 2.500 UI/kg al día o 10.000 UI/kg al día o 4.000 UI/g grasa dieta |
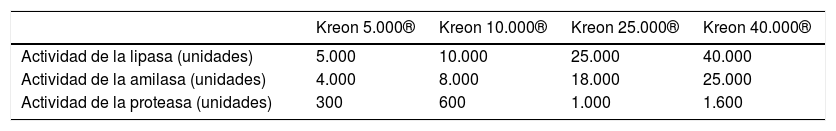
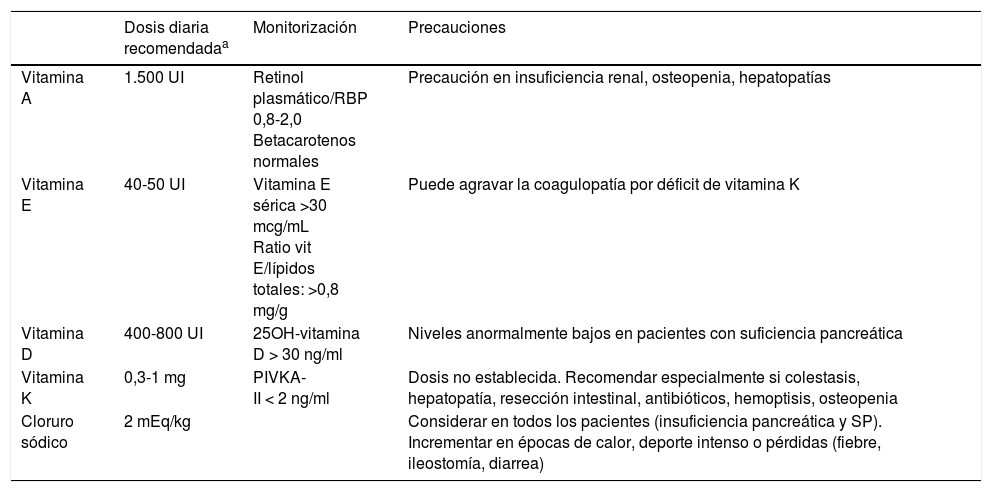
Existen diversas formulaciones (tabla 4). Las enzimas deben tomarse con cada comida. Se recomienda administrarlas con agua o alimentos ácidos como zumo o compota de manzana para evitar así que la cubierta de las microcápsulas se abra antes de tiempo20. La suplementación sistemática con vitaminas liposolubles (A, D, E y, en menor grado, K) está indicada en todos los lactantes con IP (tabla 5)16.
Actuales formulaciones de tratamiento sustitutivo enzimático en España
| Kreon 5.000® | Kreon 10.000® | Kreon 25.000® | Kreon 40.000® | |
|---|---|---|---|---|
| Actividad de la lipasa (unidades) | 5.000 | 10.000 | 25.000 | 40.000 |
| Actividad de la amilasa (unidades) | 4.000 | 8.000 | 18.000 | 25.000 |
| Actividad de la proteasa (unidades) | 300 | 600 | 1.000 | 1.600 |
Dosis recomendada de vitaminas liposolubles y minerales en lactantes menores de un año con insuficiencia pancreática
| Dosis diaria recomendadaa | Monitorización | Precauciones | |
|---|---|---|---|
| Vitamina A | 1.500 UI | Retinol plasmático/RBP 0,8-2,0 Betacarotenos normales | Precaución en insuficiencia renal, osteopenia, hepatopatías |
| Vitamina E | 40-50 UI | Vitamina E sérica >30 mcg/mL Ratio vit E/lípidos totales: >0,8 mg/g | Puede agravar la coagulopatía por déficit de vitamina K |
| Vitamina D | 400-800 UI | 25OH-vitamina D > 30 ng/ml | Niveles anormalmente bajos en pacientes con suficiencia pancreática |
| Vitamina K | 0,3-1 mg | PIVKA-II < 2 ng/ml | Dosis no establecida. Recomendar especialmente si colestasis, hepatopatía, resección intestinal, antibióticos, hemoptisis, osteopenia |
| Cloruro sódico | 2 mEq/kg | Considerar en todos los pacientes (insuficiencia pancreática y SP). Incrementar en épocas de calor, deporte intenso o pérdidas (fiebre, ileostomía, diarrea) |
Basadas en las recomendaciones de la European Cystic Fibrosis Society16.
Retinol sérico (mg/dL)/ retinol binding protein (RBP) (mg/dL)×0,0734.
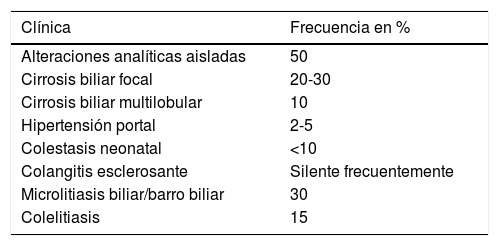
El hígado puede afectarse de forma heterogénea (tabla 6).
Afectación hepática en la fibrosis quística
| Clínica | Frecuencia en % |
|---|---|
| Alteraciones analíticas aisladas | 50 |
| Cirrosis biliar focal | 20-30 |
| Cirrosis biliar multilobular | 10 |
| Hipertensión portal | 2-5 |
| Colestasis neonatal | <10 |
| Colangitis esclerosante | Silente frecuentemente |
| Microlitiasis biliar/barro biliar | 30 |
| Colelitiasis | 15 |
No debemos olvidar la recomendación de suplementar cloruro sódico en periodos de calor.
RespiratorioTécnicas que mejoran el aclaramiento mucociliarLas secreciones deshidratadas de estos pacientes ocasionan disfunción en el aclaramiento mucociliar, lo que da lugar a un círculo vicioso de inflamación e infección pulmonar crónica, por lo que el inicio de la fisioterapia respiratoria debe ser precoz21. Existen varias técnicas de fisioterapia respiratoria a estas edades: percusión y drenaje postural, y compresión torácica de alta frecuencia. La evidencia actual no permite afirmar qué método es mejor.
El tratamiento con DNasa reduce la viscosidad del moco y facilita el aclaramiento mucociliar. Se administran 2,5ml nebulizados/día (previo broncodilatador), en mayores de 6 años; en preescolares, solo se considerará en determinados casos.
El suero salino hipertónico actúa como agente osmótico restaurando el líquido de la superficie de la vía aérea. La nebulización de 4ml de este suero al 7%, 2 veces al día (previo broncodilatador) en pacientes mayores de 6 años mostró eficacia en la función pulmonar y en las exacerbaciones22,23.
Tratamiento de la inflamaciónLa azitromicina modula la producción de citocinas e indirectamente interviene en la formación de biofilms. En los casos en los que se indique (por ejemplo, en cultivos persistentes positivos a Pseudomonas), se debe administrar 3 veces por semana, a la dosis habitual, vigilando la función hepática y teniendo en cuenta que su continua coadministración con tobramicina inhalada podría disminuir el efecto de esta última24.
Infecciones respiratoriasPrevención de la infecciónLa realización del CN de FQ permite instruir a la familia y al niño desde pequeño sobre la importancia de la higiene de manos en el entorno familiar25.
En la consulta se aconseja minimizar el tiempo de espera, utilizar sus propios juguetes y usar mascarilla26. Actualmente es unánime la recomendación de una segregación universal a todos estos pacientes. Son recomendables las vacunas reglamentarias del calendario vacunal, incluida la de la gripe (en mayores de 6 meses) y evitar la asistencia a guardería.
Tratamiento de las primoinfecciones bacterianasLa ocupación por secreciones viscosas de las vías respiratorias, característica de estos pacientes, produce de forma secundaria infecciones recurrentes por microorganismos. Los más frecuentes son:
Staphylococcus aureusA)Staphylococcus aureussensible a meticilina
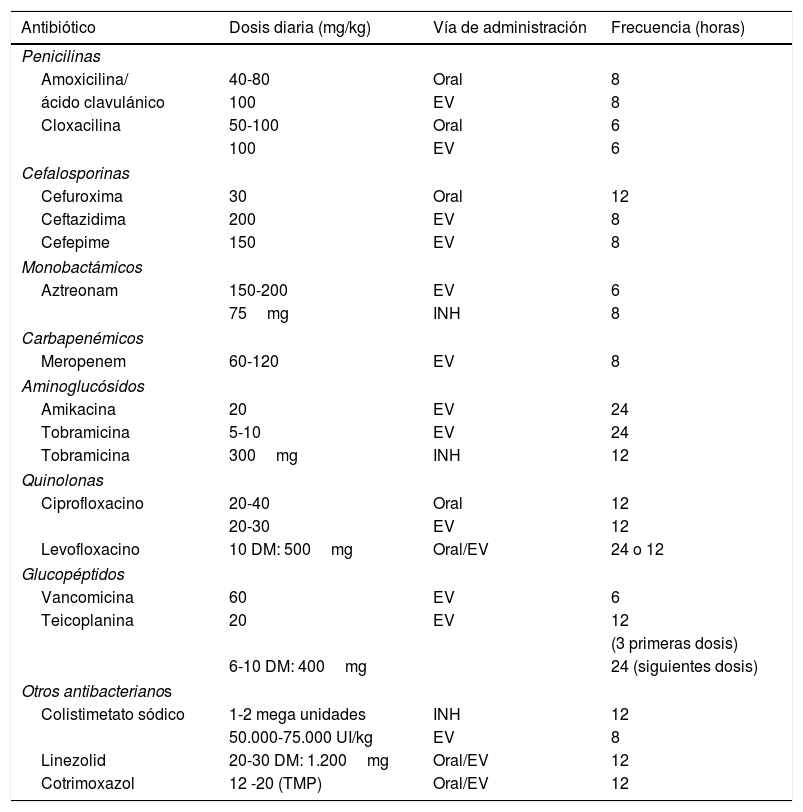
No hay evidencias suficientes a favor o en contra para recomendar la erradicación del S. aureus o el uso de profilaxis27. Los fármacos más utilizados son: amoxicilina-clavulánico, cefadroxilo y trimetropim-sulfametoxazol (TMP-SMX) por vía oral durante 14-21 días. Se aconseja realizar cultivo de secreciones una semana después de finalizar el tratamiento (tabla 7).
Antibióticos empleados en la afectación respiratoria en la fibrosis quística
| Antibiótico | Dosis diaria (mg/kg) | Vía de administración | Frecuencia (horas) |
|---|---|---|---|
| Penicilinas | |||
| Amoxicilina/ | 40-80 | Oral | 8 |
| ácido clavulánico | 100 | EV | 8 |
| Cloxacilina | 50-100 | Oral | 6 |
| 100 | EV | 6 | |
| Cefalosporinas | |||
| Cefuroxima | 30 | Oral | 12 |
| Ceftazidima | 200 | EV | 8 |
| Cefepime | 150 | EV | 8 |
| Monobactámicos | |||
| Aztreonam | 150-200 | EV | 6 |
| 75mg | INH | 8 | |
| Carbapenémicos | |||
| Meropenem | 60-120 | EV | 8 |
| Aminoglucósidos | |||
| Amikacina | 20 | EV | 24 |
| Tobramicina | 5-10 | EV | 24 |
| Tobramicina | 300mg | INH | 12 |
| Quinolonas | |||
| Ciprofloxacino | 20-40 | Oral | 12 |
| 20-30 | EV | 12 | |
| Levofloxacino | 10 DM: 500mg | Oral/EV | 24 o 12 |
| Glucopéptidos | |||
| Vancomicina | 60 | EV | 6 |
| Teicoplanina | 20 | EV | 12 |
| (3 primeras dosis) | |||
| 6-10 DM: 400mg | 24 (siguientes dosis) | ||
| Otros antibacterianos | |||
| Colistimetato sódico | 1-2 mega unidades | INH | 12 |
| 50.000-75.000 UI/kg | EV | 8 | |
| Linezolid | 20-30 DM: 1.200mg | Oral/EV | 12 |
| Cotrimoxazol | 12 -20 (TMP) | Oral/EV | 12 |
DM: dosis máxima; EV: endovenoso; INH: inhalado.
B)Staphylococcus aureusresistente a meticilina
En pacientes asintomáticos o con clínica leve: TMP-SMX, linezolid, 2-4 semanas28. En pacientes con clínica grave se recomienda tratamiento con antibióticos intravenosos durante 2-3 semanas con vancomicina o teicoplanina. En el caso de cronicidad, una opción es usar vancomicina nebulizada durante 2-3 semanas.
Haemophilus influenzaeSi el paciente tiene clínica, la amoxicilina-clavulánico y TMP-SMX (2-4 semanas) son los antibióticos más utilizados.
Pseudomonas aeruginosaEl tratamiento erradicador debe iniciarse tras la primera detección de P. aeruginosa. De los protocolos de erradicación publicados ninguno demostró superioridad. Según el consenso español, el manejo del primer aislamiento de P. aeruginosa29consiste en:
1)Sin clínica: ciprofloxacino oral 2-4 semanas + antibióticos nebulizados: colistina diaria (máximo 3-6 meses), o 1-3 ciclos on-off de tobramicina o aztreonam (máximo 6 meses).
2) Con clínica: antibioterapia sistémica + inhalada.
- Leve: ciprofloxacino oral 2-4 semanas.
- Moderada-grave: antibióticos intravenosos: ceftazidima + tobramicina (2-3 semanas).
Se aconseja realizar cultivo de secreciones una semana después de finalizar el tratamiento: si es negativo, mantener colistina diaria (máximo 3-6 meses), o 1-3 ciclos on-off de tobramicina o aztreonam (máximo 6 meses).
Tratamiento de las exacerbaciones respiratoriasSe necesita contar con una definición aceptada de exacerbación respiratoria en lactantes y preescolares con FQ. Algunos grupos la definen como cualquier cambio en la clínica respiratoria habitual.
A mayor número de exacerbaciones en los 2primeros años de vida, mayor progresión de la enfermedad30. Ante una infección respiratoria se debe plantear inmediatamente un cultivo de secreciones respiratorias e instaurar tratamiento antibiótico en función de la edad, características clínicas, análisis microbiológicos previos15 y con una duración de 2-4 semanas.
Si, a pesar de un tratamiento adecuado, no existe mejoría, es posible que la infección sea debida a microorganismos resistentes a los antibióticos pautados (evaluar en este caso la necesidad de practicar una fibrobroncoscopia y un lavado broncoalveolar), o a la existencia de otras enfermedades como reflujo gastroesofágico, alteraciones en la deglución, etc.14,15,30. No se debe olvidar en ningún momento la fisioterapia respiratoria y una nutrición e hidratación adecuadas.
Tratamiento del defecto básico con moduladores de la proteína CFTREn la actualidad se dispone de tratamientos dirigidos al defecto subyacente de la enfermedad. Estas nuevas terapias pretenden mejorar la función del CFTR con compuestos que actúan sobre la proteína, ya sea con correctores (clase II) o con potenciadores (clases III y IV). Ivacaftor (Kalydeco®) es un potenciador que demostró eficacia (aumento de la función pulmonar y el índice de masa corporal, y redujo el cloro en sudor y hasta un 52% las exacerbaciones respiratorias) y seguridad en los pacientes con mutaciones de clase iii31. La dosis es un comprimido de 150mg cada 12 h coincidiendo con la ingesta de comidas grasas. En España está aprobado a partir de los 6 años y en EE. UU. a partir de los 12 meses.
La combinación de un corrector y un potenciador del CFTR, lumacaftor+ivacaftor (Orkambi®), es una nueva estrategia para los homocigotos F508del (clase II) y demostró efectos beneficiosos sobre la función pulmonar, número de exacerbaciones y calidad de vida32. La posología es de 2 comprimidos cada 12 h. Está autorizado en EE. UU. a partir de los 2 años, pero aún no está disponible en España.
Seguimiento del recién nacido diagnosticado de fibrosis quística por cribado neonatalRevisiones programadas y revisión anualEn la tabla 5 se muestra el calendario de visitas programadas aconsejables en pacientes asintomáticos o con estabilidad clínica. Este calendario debe, no obstante, individualizarse en función de la sintomatología y necesidades familiares14.
Ante alguna eventualidad o duda, las familias deben poder contactar con el equipo y conocer el protocolo de actuación. Ante cualquier cambio en los síntomas que pudiera significar una exacerbación pulmonar, deben poder recibir asistencia para realización de cultivo de secreciones y valorar tratamiento antibiótico empírico33.
Exploraciones complementariasEn la actualidad se recomienda una radiografía de tórax cuando se sospeche una complicación, o en una exacerbación moderada o grave.
La tomografía computarizada de tórax(TC) es la prueba de imagen patrón oro para detectar precozmente las primeras alteraciones (atrapamiento aéreo, bronquiectasias) y su progresión: es más sensible que la radiografía de tórax.
Las bronquiectasias pueden encontrarse hasta en un 50% a la edad de 5 años34,35. El protocolo de la TC para la FQ debe incluir cortes inspiratorios y espiratorios con bajas dosis de radiación. En la mayoría de los centros se realiza una TC de rutina a partir de los 2 años (sin anestesia) y cada 2años coincidiendo con el control anual, mientras que en otros se efectúa a partir de los 4-5 años con controles más esporádicos.
Función pulmonar del preescolarLa espirometría forzada es la técnica más utilizada. El volumen espiratorio forzado en el primer segundo (FEV1) es útil para definir la presencia de una exacerbación y su respuesta al tratamiento. Sin embargo, se ha comprobado que, aun teniendo alteraciones estructurales, los preescolares pueden mantener una función pulmonar normal.
Índice de aclaramiento pulmonarEl índice de aclaramiento pulmonar es una prueba nueva y muy sensible que detecta alteraciones más precozmente que la espirometría, y evalúa la heterogeneidad en la distribución de la ventilación36. De momento se utiliza en ensayos clínicos37,38.
ConclusionesEl CN de FQ ofrece una oportunidad única para el diagnóstico precoz que ha demostrado mejorar el estado nutricional, la función pulmonar y la supervivencia, y que disminuye los ingresos hospitalarios, todo ello con la posibilidad de reducir el coste terapéutico. Está ampliamente implementado en el mundo y en España. Solo se obtendrá un efecto beneficioso si se cumplen por completo las normas de atención seguida de los standards of care, por lo que creemos imprescindible publicar estas normas de diagnóstico y seguimiento de los pacientes con FQ diagnosticados a través del CN.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
María José Alonso: Instituto de Biología y Genética Molecular (IBGM), Universidad de Valladolid, Valladolid.
Marina Álvarez: Unidad de Gastroenterología Pediátrica, Hepatología y Soporte Nutricional, Hospital Universitari Vall d́Hebron, Barcelona.
Anselmo Andrés Martín: Sección de Neumología Pediátrica, Servicio y UGC de Pediatría, Hospital Universitario Virgen Macarena, Sevilla.
María Isabel Barrio Gómez de Agüero: Unidad de Neumología Pediátrica y Fibrosis Quística, Hospital Universitario La Paz, Madrid
María Jesús Cabero Pérez: Unidad de Neumología Infantil y Fibrosis Quística, Hospital Marqués de Valdecilla, Santander.
Pilar Caro Aguilera: Unidad de Neumología Infantil y Fibrosis Quística, Hospital Materno-Infantil Universitario de Málaga, Málaga.
María Cols Roig: Unidad de Neumología Infantil y Fibrosis Quística, Servicio de Pediatría, Hospital Sant Joan de Déu, Barcelona.
Isidoro Cortell Aznar-Pérez: Servicio de Neumología y Alergia Pediátricas, Hospital Universitario La Fe, Valencia.
Jordi Costa Colomer: Unidad de Neumología Infantil y Fibrosis Quística, Servicio de Pediatría, Hospital Sant Joan de Déu, Barcelona.
Isabel Delgado Pecellín: Unidad de Neumología Infantil y Fibrosis Quística, Hospital Universitario Virgen del Rocío, Sevilla.
Amparo Escribano Montaner: Unidad de Neumología Infantil y Fibrosis Quística, Hospital Clínico Universitario de Valencia, Universitat de València, Valencia.
Joan Figuerola Mulet: Unidad de Neumología Infantil y Fibrosis Quística, Hospital Universitario Son Espases, Palma de Mallorca.
Gloria García Hernández: Unidad de Neumología Infantil y Fibrosis Quística, Hospital Universitario 12 de Octubre, Madrid.
Pilar Guayarte: Unidad de Gastroenterología Pediátrica, Hospital Universitario Parc Taulí, Sabadell.
David Gil Ortega: Unidad de Gastroenterología, Hepatología y Nutrición Pediátrica, Hospital Universitario Virgen de la Arrixaca, Murcia.
David Gómez Pastrana: Unidad de Neumología Infantil, Hospital de Jerez, Jerez de la Frontera.
Adelaida Lamas Ferreiro: Unidad de Fibrosis Quística, Hospital Universitario Ramón y Cajal, Instituto Ramón y Cajal de Investigación Sanitaria (IRYCIS), Madrid.
José Luis Marín Soria: Laboratorio de Cribado Neonatal de Cataluña, Hospital Clinic, Barcelona.
Carlos Martín de Vicente: Unidad de Neumología Pediátrica y Fibrosis Quística, Hospital Universitario Miguel Servet, Zaragoza.
Martín Navarro Merino: Sección de Neumología Pediátrica, Servicio y UGC de Pediatría, Hospital Universitario Virgen Macarena, Sevilla.
Concepción Oliva Hernández: Hospital Universitario Nuestra Señora de la Candelaria, Santa Cruz de Tenerife.
Javier Pérez Frías: Unidad de Neumología Infantil y Fibrosis Quística, Hospital Materno-Infantil Universitario de Málaga, Málaga.
Estela Pérez Ruiz: Unidad de Neumología Pediátrica y Fibrosis Quística, Hospital Materno-Infantil Universitario de Málaga, Málaga.
Sandra Rovira Amigo: Unidad de Neumología Pediátrica y Fibrosis Quística, Hospital Vall dH́ebron, Barcelona.
Antonio Salcedo Posadas: Unidad de Fibrosis Quística, Hospital Universitario del Niño Jesús-Hospital Universitario Gregorio Marañón, Madrid.
Manuel Sánchez-Solís: Unidad de Neumología Infantil y Fibrosis Quística, Hospital Universitario Virgen de la Arrixaca, Murcia.
Josep Sirvent Gómez: Unidad de Neumología Pediátrica, Hospital Materno Infantil, Complexo Hospitalario Universitario A Coruña, La Coruña.
Carlos Vázquez Cordero: Unidad de Neumología Pediátrica y Fibrosis Quística, Hospital Universitario Cruces, Bilbao.
José Ramón Villa Asensi: Unidad de Neumología y Fibrosis Quística, Hospital Universitario del Niño Jesús, Madrid.
