






El Programa de cribado o detección precoz del hipotiroidismo congénito (HC) es uno de los mayores avances logrados en Pediatría. Las hormonas tiroideas son imprescindibles para el desarrollo y la maduración cerebral, que continúan en la etapa neonatal. El hipotiroidismo de comienzo en los primeros meses de vida origina lesiones irreversibles en el sistema nervioso central y es una de las causas más frecuentes y evitables de retraso mental. El diagnóstico clínico es tardío, por lo que requiere estudio analítico para poder efectuar el tratamiento adecuado.
Este artículo actualiza los objetivos, los procedimientos diagnósticos, las pruebas imprescindibles y complementarias requeridas, la etiología y los diagnósticos diferenciales en esta patología. Con especial énfasis en los requerimientos de los centros de seguimiento para protocolizar los resultados del tratamiento con L-tiroxina administrada de forma inmediata al diagnóstico y a las dosis que eviten fases de infra o supradosificación que pueden alterar diversos aspectos del desarrollo cognitivo. La revaluación de etiología permanente vs. transitoria se recomienda siempre después de los 3años de edad.
La relevancia de este programa precisa su difusión a todas las áreas de pediatría. El objetivo principal, evitar el daño cerebral en estos pacientes, se ha logrado y es además altamente beneficioso desde el punto de vista económico.
Otros aspectos para optimizar los resultados cognitivos con todos los controles periódicos necesarios y lograr la inclusión del diagnóstico del HC central, precisan implementar los recursos de los centros de seguimiento y continuar avanzando según los conocimientos actuales.
The screening program of congenital hypothyroidism (CH) is probably one of the best achievements in paediatrics. Thyroid hormones are essential for brain development and brain maturation that continue through the neonatal period. Hypothyroidism that begins in the first months of life causes irreversible damage to the central nervous system, and is one of the most frequent and preventable causes of mental retardation. As children with congenital hypothyroidism are born with a normal appearance, analytical studies are required to immediately start the appropriate therapy.
This article analyses the aims, diagnostic procedures, tests required, aetiology, and differential diagnosis in this disorder. Especially relevant is to perform frequent monitoring to ensure dose adjustments of L-Thyroxine therapy, avoiding infra- or supra-dosing that negatively affects neurosensory functions. Re-evaluation of the aetiology permanent vs transient hypothyroidism is always recommended after 3years of chronological age.
The relevance of this screening program should be widely discussed in paediatrics. The main objective is to avoid cerebral damage in these patients, and has been highly successful and economically beneficial.
Other aspects are required to optimise patient outcomes, to perform all the controls according to the recommendations and to include, in the near future, the diagnosis of central hypothyroidism. Implementation of this program is necessary to progress in accordance with current scientific knowledge.
El hipotiroidismo se caracteriza por la situación clínica y analítica resultante de la disminución de la actividad biológica de las hormonas tiroideas a nivel tisular; en la mayoría de los casos, coincide con un descenso de los niveles plasmáticos de hormonas tiroideas y elevación de tirotropina hipofisaria (TSH).
El hipotiroidismo congénito (HC) y neonatal comprende un grupo heterogéneo de alteraciones tiroideas que producen hipofunción tiroidea, detectable ya en la primera etapa de la vida del recién nacido1,2.
Las hormonas tiroideas son imprescindibles para lograr el desarrollo y la maduración cerebral normales. El hipotiroidismo de comienzo en los primeros meses de vida originará lesiones irreversibles en el sistema nervioso central. El HC y neonatal debe de ser diagnosticado y tratado de forma urgente, porque es una causa frecuente y evitable de retraso mental3-6.
Los síntomas clínicos son inespecíficos, progresan en relación directa con el tiempo transcurrido y la intensidad del hipotiroidismo. En el primer mes de vida, etapa en la que debe de iniciarse el tratamiento, solamente un 5% de los niños afectos serían diagnosticables clínicamente.
El primer programa de cribado neonatal de HC se realizó en Quebec en 19741. Actualmente, la mayoría de los países desarrollados ofrecen esta cobertura, no así los países en desarrollo7.
Estos programas han permitido conocer que el HC es muy frecuente y han evitado, en la gran mayoría de los casos, el daño cerebral con retraso mental permanente que se observaba anteriormente en estos niños.
La prevalencia del HC primario, por afectación de la glándula tiroides, es, en series internacionales, de 1:2.000 recién nacidos y coincide con los datos publicados de España7,8.
Objetivo del programa de cribado neonatal de hipotiroidismo congénitoEl Programa de Detección Precoz del Hipotiroidismo Congénito, de interés primordial en Salud Pública y Medicina Preventiva, está incluido en los programas de cribado neonatal.
El objetivo principal es la detección y el tratamiento del HC severo y permanente. La detección del HC leve permanente o transitorio sería objetivo secundario.
En todos ellos se evita el daño neurológico y se reducen la morbilidad, la mortalidad y las posibles discapacidades asociadas a dichas enfermedades.
Es importante resaltar que las pruebas de cribado neonatal no son procedimientos de diagnóstico. Los pacientes que presenten un resultado positivo requerirán procedimientos diagnósticos posteriores, para ello se debe contar con el apoyo de clínicos especializados en su diagnóstico y tratamiento (centros clínicos de seguimiento [CCS]). Los CCS deberían reunir determinadas condiciones (tablas 1 y 2). Su objetivo es confirmar el diagnóstico de HC e iniciar rápidamente el tratamiento o excluir dicha enfermedad8,9.
Objetivos del Centro Clínico de Seguimiento de hipotiroidismo congénito
| Confirmar el diagnóstico de HC mediante la historia clínica, exploración y datos analíticos (bioquímicos y de imagen) necesarios, con disponibilidad inmediata de resultados |
| Informar y tranquilizar a la familia; explicar ventajas de la detección precoz |
| Iniciar el tratamiento urgente (L-tiroxina) y optimizarlo en controles periódicos para normalizar la función tiroidea |
| Lograr un desarrollo neurológico y psicomotor y cociente intelectual en límites normales y evitar comorbilidades |
| Diagnosticar la causa (HC permanente/transitorio) |
| Diagnosticar otras alteraciones congénitas que pudieran asociarse |
| Mantener una información bidireccional con el Centro de Diagnóstico para poder evaluar los resultados y eficacia del Programa |
| Establecer una relación directa con el pediatra del niño para poder realizar un tratamiento integral |
Requisitos del Centro Clínico de Seguimiento de hipotiroidismo congénito
| Especialistas en Endocrinología Pediátrica con experiencia en esta enfermedad y disponibilidad plena |
| Servicio de Bioquímica, para determinaciones analíticas de confirmación diagnóstica rápida y controles posteriores |
| Servicio de Medicina Nuclear (gammagrafía tiroidea inicial urgente) |
| Servicio de Radiología pediátrica (ecografía tiroidea, maduración ósea) |
| Servicio de Psicología para evaluaciones |
| Hospitalización, Neonatología |
| Relación con el pediatra del paciente |
| Comunicación directa personal con el Centro de Diagnóstico para evaluar resultados y eficacia del programa |
| Relación periódica con la Subdirección General de Promoción de la Salud y Prevención de la Consejería de Sanidad u organismo responsable del Programa |
| Reuniones de Comité de Expertos para evaluar y mejorar los resultados del programa |
El cribado neonatal no debe identificarse solo con un procedimiento de laboratorio, sino con una actividad multidisciplinar cuya coordinación con el sistema sanitario asistencial resulta imprescindible para asegurar su eficacia y eficiencia.
Procedimiento en el cribado de hipotiroidismo congénitoMedición de TSHLa toma de muestras se planifica de forma que alcance una cobertura del 100% de los recién nacidos y el tratamiento precoz del 100% de los casos detectados.
La detección precoz del hipotiroidismo congénito primario se lleva a cabo por medición de TSH a las 48 h de vida, evitando el aumento fisiológico inicial de esta hormona.
La obtención de muestra de sangre capilar por punción del talón del recién nacido se realiza sobre papel standard absorbente, precisa personal sanitario entrenado para optimizar los resultados8.
La TSH se analiza por inmunofluorescencia (DELFIA®, Laboratorios Perkinelmer). El punto de corte, por encima del cual existe sospecha de presentar la enfermedad, está establecido en > 7-10UI/ml. Se realiza, de forma complementaria, la medición de tiroxina total (T4T) cuando la TSH es superior al punto de corte establecido.
Actualmente, en España en todos los centros de cribado de HC se analiza TSH y solo en algunas comunidades autónomas (vasca, Navarra y Cantabria) se determinan inicial y simultáneamente TSH y T4T.
El hipotiroidismo central (secundario o terciario) no es detectado en los programas que analizan únicamente TSH10,11.
Confirmación diagnóstica del hipotiroidismo congénitoEl centro de cribado, ante un resultado analítico positivo, contacta de forma urgente al paciente identificado y se remite inmediatamente al CCS, donde se realizará la confirmación de hipotiroidismo y el estudio etiológico de la alteración tiroidea, sin demorar el comienzo del tratamiento12-17.
La etiología del hipotiroidismo congénito es multifactorial, el 95% de los casos es debido a hipotiroidismo primario (alteración de la glándula tiroides) y más infrecuente el de origen central.
El hipotiroidismo congénito y neonatal aparece ya en el momento del nacimiento. Según la causa, puede ser permanente, precisa tratamiento durante toda la vida; o transitorio, reversible espontáneamente al desaparecer la causa que lo originó (p. ej., un exceso de yodo o presencia de anticuerpos antitiroideos maternos). Sin embargo, todos los casos requieren tratamiento para normalizar la función tiroidea durante la época del desarrollo cerebral, siendo ineficaz si comienza posteriormente («efecto ventana»)4.
La causa más frecuente de HC primario permanente es la disgenesia tiroidea (85-90%), la mayoría de los casos ectopias tiroideas (60-65%) y en segundo lugar agenesia o atireosis (35-40%). Las dishormonogénesis (tabla 3) corresponden a un 10% de los casos. En algunos pacientes con disgenesia tiroidea se han identificado mutaciones específicas en el gen TTF-2 y en factores de transcripción de génesis tiroidea.
Etiología del hipotiroidismo congénito. Dishormonogénesis tiroidea
| 1. Defecto del atrapamiento o transporte de yodo |
| 2. Alteraciones de la organificación |
| Mutación del gen de peroxidasa |
| Alteraciones de la fuente de peróxido de hidrógeno |
| Alteraciones aceptores de yodo |
| Síndrome de Pendred |
| 3. Déficit del acoplamiento de las yodotironinas |
| Síndrome de Hollander |
| 4. Déficit de la deshalogenasa de yodotirosinas |
| 5. Yodoproteínas anómalas |
| 6. Alteraciones de la síntesis de tiroglobulina |
| Mutación del gen de Tg |
| Reducción de TgmRNA |
| Proteína Tg truncada |
| Hiposialización de Tg |
| 7. Insensibilidad a TSH |
La etiología del hipotiroidismo es en algunos casos desconocida; numerosos genes pueden estar implicados en la etiología multifactorial del hipotiroidismo congénito primario17-24.
Comunicación del diagnósticoEs un eslabón primordial en el diagnóstico y el tratamiento del HC. Busca que los padres comprendan el beneficio que supone el diagnóstico precoz para prevenir el daño cerebral, aprendan a administrar correctamente la mediación, mantengan una buena adherencia al tratamiento y realicen un adecuado seguimiento el tiempo que dure el HC (meses, años, o toda la vida). La información inicial la debe realizar personal experto, asegurándose de que los padres comprenden perfectamente la información.
Actuación en el centro clínico de seguimiento de hipotiroidismo congénitoLa pauta de actuación ante un resultado positivo en el cribado neonatal se expone en la figura 1.

En todos los casos se realiza una anamnesis personal y familiar, con especial incidencia en posibles antecedentes de ingesta de fármacos o utilización de compuestos yodados, historia de enfermedad tiroidea familiar, sobre todo materna, y en síntomas de hipofunción tiroidea.
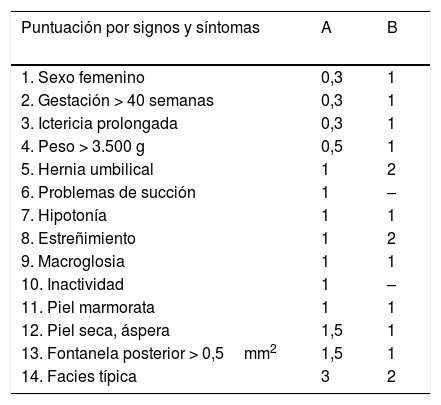
La exploración clínica aporta los síntomas o signos del hipotiroidismo (tabla 4).
Signos y síntomas clínicos en hipotiroidismo congénito. Índice de hipotiroidismo congénito
| Puntuación por signos y síntomas | A | B |
|---|---|---|
| 1. Sexo femenino | 0,3 | 1 |
| 2. Gestación > 40 semanas | 0,3 | 1 |
| 3. Ictericia prolongada | 0,3 | 1 |
| 4. Peso > 3.500 g | 0,5 | 1 |
| 5. Hernia umbilical | 1 | 2 |
| 6. Problemas de succión | 1 | – |
| 7. Hipotonía | 1 | 1 |
| 8. Estreñimiento | 1 | 2 |
| 9. Macroglosia | 1 | 1 |
| 10. Inactividad | 1 | – |
| 11. Piel marmorata | 1 | 1 |
| 12. Piel seca, áspera | 1,5 | 1 |
| 13. Fontanela posterior > 0,5mm2 | 1,5 | 1 |
| 14. Facies típica | 3 | 2 |
Puntuación clínica indicativa de hipotiroidismo congénito: superior a 4 puntos.
Fuente: A, Letarte et al., 198012.
Los resultados obtenidos en la muestra de sangre en papel de filtro son confirmados mediante analítica en sangre venosa (TSH, T4L). En el hipotiroidismo primario las cifras elevadas de TSH se acompañan de valores disminuidos de T4L. La determinación de tiroglobulina informa de la presencia de glándula tiroides. La presencia de anticuerpos antitiroideos apoya el diagnóstico de bloqueo transitorio autoinmune del tiroides. La yoduria elevada, superior a 200 μg/l indica exposición a exceso de yodo con bloqueo de la función tiroidea.
Las pruebas de imagen, ecografía y gammagrafía tiroidea son de gran utilidad para determinar la causa del HC. El diagnóstico de disgenesia es definitivo, mientras que un tiroides eutópico con captación normal puede indicar HC transitorio.
La ecografía tiroidea permite verificar la presencia de tejido tiroideo en el cuello. Puede diferenciar la disgenesia tiroidea (agenesia o ectopia) de la dishormonogénesis, que se representa con tiroides de localización normal. Sin embargo, la interpretación de los resultados de la ecografía tiroidea en el neonato requiere una alta especialización.
La gammagrafía tiroidea con 123I o 99Tc localiza la glándula tiroides, define su tamaño y establece el diagnóstico definitivo de agenesia, ectopia, hemiagenesia o hipoplasia. Si no se visualiza el tiroides gammagráficamente y las cifras de tiroglobulina son elevadas, será útil realizar una ecografía, ya que la glándula puede estar bloqueada por la presencia de anticuerpos bloqueantes del tiroides (enfermedad tiroidea autoinmune materna) o tratarse de un defecto el atrapamiento del yodo en dishormonogénesis (NIS), una alteración genética de TSH-β o mutaciones inactivantes del receptor de TSH. En casos de bloqueo parcial, el tiroides puede estar eutópico y ser de tamaño normal.
Otros estudios (no imprescindibles en el diagnóstico inicial)Potenciales auditivos evocados: la relación entre la hipofunción tiroidea y la sordera es bien conocida. Algunas dishormonogénesis (síndrome de Pendred, síndrome de Hollander) deben sospecharse si existe sordera familiar.
Los pacientes con HC pueden presentar anomalías congénitas en mayor frecuencia que la población general, en especial alteraciones cardiacas; por ello es conveniente realizar un estudio cardiológico, La evaluación inicial por neuropediatría también es de gran utilidad, ya que pueden existir otras enfermedades que produzcan retraso del desarrollo psicomotor.
Diagnóstico diferencialSi en un recién nacido se detecta HC mediante cribado neonatal y en el análisis realizado en el CCS se confirma mediante el estudio de función tiroidea, no se debe plantear ningún diagnóstico diferencial y menos aún posponer el inicio del tratamiento para definir exhaustivamente su etiología. El diagnóstico definitivo se podrá esclarecer en muchos casos en el replanteamiento a realizar a los 3años de edad cronológica, y en otros, sobre todo transitorios, persistirá la duda sobre su etiología.
El pediatra ha de cerciorarse de la normalidad de la función tiroidea en los recién nacidos procedentes de países donde no existe este cribado.
Existen situaciones especiales en las que se recomienda realizar un protocolo de muestras seriadas de TSH a las 2 y 4 semanas de vida, por posible elevación tardía de TSH o por necesidad de determinar T4 y TSH (tabla 5)25-28.
Situaciones especiales en la detección precoz de hipotiroidismo congénito
| Recién nacidos con <30 semanas de gestación |
| Recién nacidos con peso igual o inferior a 1.500 g |
| Utilización de antisépticos yodados en el parto o en el recién nacido (cesáreas, cirugía mayor) |
| Realización de técnicas radiológicas con contrastes yodados |
| Inserción de catéteres de silástico, cateterismos cardiacos |
| Neonatos con síndrome de Down |
| Partos múltiples; particularmente gemelos del mismo sexo |
| (posibilidad de transfusión feto-fetal) |
| Neonatos ingresados en Unidades de Cuidados Intensivos |
| Muestras postransfusionales |
Fuente: Rodríguez Sánchez et al.15.
En el hipotiroidismo congénito y neonatal el daño cerebral depende directamente del tiempo transcurrido desde el comienzo del hipotiroidismo y el comienzo del tratamiento. El tratamiento debe iniciarse lo antes posible; es recomendable realizarlo antes de los 15días de edad. La edad de comienzo del tratamiento debe coincidir con el día del diagnóstico y no debe retrasarse el comienzo del tratamiento para realizar pruebas complementarias de localización diagnóstica.
El fármaco de elección es la L-tiroxina sintética por vía oral, administrada 30 min antes de una toma, cada 24 h, a dosis de 10-15 μg/kg/día29-33.
Los preparados genéricos de L-tiroxina no presentan la misma biodisponibilidad34. Las fórmulas magistrales liquidas de L-tiroxina no son estables. La absorción se ve modificada si se ingieren preparados con soja, hierro, calcio, hidróxido de aluminio, omeprazol, fibra, sucralfato o resinas.
Después de comenzar el tratamiento con L-tiroxina se deben realizar controles clínicos y analíticos frecuentes para optimización terapéutica. Recomendamos una segunda visita a los 2 días para confirmar la aceptación del diagnóstico y el tratamiento, una tercera exploración a los 15 días cuando se realizan determinación de TSH y T4L; después mensualmente durante los primeros 6meses de vida; posteriormente cada 2meses hasta el año de edad y cada 3-4 meses hasta su reevaluación a los 3 años de edad.
En cada visita se debe realizar una exploración física completa y análisis de niveles de T4L y TSH. El objetivo del tratamiento es normalizar rápidamente los niveles de T4L y TSH. T4L debe permanecer en límites altos de normalidad, evitando tanto hipo como hipertiroidismo subclínico35. Si se produce una modificación en la dosis de L-tiroxina se efectuará un control de TSH y T4L a las 4 semanas.
Diagnóstico definitivo del hipotiroidismo congénitoEl tratamiento del niño detectado en los programas de detección precoz de HC debe mantenerse sin interrupción durante los 3primeros años de vida, para asegurar la normofunción tiroidea hasta completar el desarrollo cerebral, tanto en los casos permanentes como transitorios. A esta edad, puede realizarse en los casos no diagnosticados la revaluación diagnóstica (fig. 2).

Los niños con diagnóstico inicial de agenesia tiroidea o tiroides ectópico, permanentes, no precisan revaluación. Se plantea la revaluación diagnóstica en los niños con HC y tiroides eutópico en el diagnóstico inicial, y en aquellos con HC y etiología no filiada36,37. También se plantea este diagnóstico en los pacientes en los que no se realizó el diagnóstico definitivo (gammagrafía) en el inicio del tratamiento. En la revaluación diagnóstica, y siempre a partir de los 3 años, se pueden seguir 2 estrategias:
Si deseamos conocer si el hipotiroidismo es permanente o transitorio, basta con disminuir la dosis de L-tiroxina que esté recibiendo a la mitad y al mes evaluar TSH y T4L: si la TSH es superior o igual a 10 mUI/l, se considera HC permanente y se reanudará el tratamiento con L-tiroxina a la dosis que le mantenía eutiroideo. En pacientes con TSH inicial muy elevada y tiroides eutópico es suficiente suspender L-tiroxina solo durante 2semanas para obtener valores elevados de TSH.
Si el objetivo es realizar un diagnóstico definitivo, se suspende el tratamiento con L-tiroxina un mes y se evalúan la T4L, la TSH, la tiroglobulina y los anticuerpos antitiroideos (antitiroglobulina, antiperoxidasa tiroidea). Cuando no se realizó antes del inicio del tratamiento, la gammagrafía tiroidea (99Tc) puede diagnosticar ectopia o posible agenesia tiroidea, confirmando esta con ecografía del tiroides. En los casos de HC permanente con tiroides eutópico la gammagrafía tiroidea con 123I permite realizar un test de descarga con perclorato para el diagnóstico de dishormonogénesis. Los estudios genéticos moleculares son de gran utilidad en el diagnóstico de HC.
Apuntan a un HC permanente con tiroides eutópico: familiares en primer grado con bocio o hipotiroidismo primario eutópico congénito, hipoplasia tiroidea en el diagnóstico inicial, necesidad de dosis altas de L-tiroxina (> 2 μg/kg/día) en el seguimiento, hiperplasia tiroidea en la revaluación.
La etiología del HC según los resultados de la reevaluación diagnóstica se expone en la tabla 3. Si se confirma una etiología permanente, la familia debe ser informada de la necesidad de mantener el tratamiento con L-tiroxina durante toda la vida, con las modificaciones de dosificación necesarias. En caso de hipotiroidismo transitorio, se discontinúa el tratamiento.
PronósticoLa detección precoz del hipotiroidismo congénito mediante programa de cribado neonatal evita el retraso mental que estos niños presentaban al realizar diagnóstico clínico, tardío. El cociente intelectual en los pacientes con HC tratados precoz y adecuadamente es normal17,18,36,38,39.
A pesar de comenzar el tratamiento precoz, en algunos casos se ha detectado disfunción cerebral mínima, que conlleva problemas de conducta, alteraciones en la comprensión del lenguaje, en la motricidad fina, discriminación perceptomotora y visuomotora, casi siempre de escasa relevancia para una vida normal. Estas alteraciones se han relacionado con la edad de inicio del tratamiento (superior a 15-21 días), dosis de L-tiroxina (generalmente inferior a la recomendada), severidad inicial del hipotiroidismo y niveles de T4L en el seguimiento inferiores o superiores a los valores recomendados. Los niveles excesivamente altos de T4L en los primeros meses se pueden asociar a falta de atención en años posteriores35.
La interrupción del tratamiento antes de los 3 años sin supervisión médica ni control posterior es una situación con graves consecuencias sobre el desarrollo neurológico.
Es importante el seguimiento para lograr no solo buen desarrollo madurativo, también evolución y crecimiento óptimos. En países donde no existen CCS de pacientes con HC se describen hasta un 25-30% de abandonos del tratamiento37,40.
Transición a la vida adultaLos niños afectados de HC han de seguirse en CCS hasta que finalicen la pubertad y alcancen talla final16,17. Posteriormente, el control debería realizarse cada 6-12 meses o más frecuentemente si se detecta un mal cumplimiento del tratamiento. El objetivo debe ser monitorizar la función tiroidea para evitar el hipo o hipertiroidismo subclínicos y su relación con problemas cardiovasculares, sobrepeso y defectos en la mineralización ósea en la vida adulta.
Solo el 70% de los adultos con HC realizan un adecuado cumplimiento del tratamiento. Esto es especialmente preocupante en el caso de mujeres con deseo concepcional o embarazadas en las que las necesidades de hormonas tiroideas aumentan. El tratamiento óptimo del hipotiroidismo materno es importante para lograr un embarazo normal con un desarrollo neurocognitivo adecuado del niño. Se recomienda que la monitorización del tratamiento sea adecuada a lo largo de toda la infancia y la vida adulta. Se precisan estrategias educacionales que mejoren la adherencia al tratamiento, especialmente en la etapa de la transición.
Problemas no resueltos en la detección precoz de hipotiroidismo congénitoAnte la sospecha clínica de hipotiroidismo, incluso con cribado normal, el pediatra debe solicitar una determinación urgente de T4L y TSH, ya que pueden existir elevaciones tardías de TSH. Otras causas infrecuentes, como el hipotiroidismo consumptivo por hiperactividad de desyodasa iii en grandes hemangiomas en el periodo posnatal, tampoco son detectadas en los programas de cribado (TSH y T4 iniciales normales)27.
El cribado que determina solo TSH no identifica los casos de HC central10,11. El HC central, aislado o asociado a hipopituitarismo, es más frecuente (1:16.000-1:20.000) de la incidencia previamente reportada (1:100.000), por lo que se aconseja ampliar el cribado con la determinación simultánea de TSH y T4T. Actualmente, se preconiza el análisis de T4 en la muestra del recién nacido; ello evitaría el diagnóstico tardío de estos pacientes, muchos de ellos asintomáticos o con clínica inespecífica.
Las alteraciones de TSH-α y TSH-β pueden manifestarse con una elevación tardía de TSH. Las alteraciones del transportador MCT8 se deben sospechar en pacientes con alteraciones neurológicas durante el primer año de vida, y cursan con función tiroidea inicial normal16,17.
ConclusionesEl Programa de Detección Precoz del HC ha sido uno de los mayores avances en prevención y salud infantil del siglo xx. La detección de estos pacientes evita el sufrimiento personal y familiar de un paciente con daño cerebral permanente. Económicamente, estos programas son de alta rentabilidad social; sin embargo, solo el 25% de la población mundial se beneficia de ellos.
Consensuar si es preciso determinar solo TSH, T4 o ambas para también detectar hipotiroidismo central, así como disminuir el nivel de corte de TSH a 5 mIU/l, para detectar hipotiroidismos más leves, precisa análisis más profundos de coste-beneficio en los que hay que trabajar evitando incrementar el número de falsos positivos.
Por todo ello, en los próximos años, estos programas deben dotarse de las infraestructuras necesarias para adecuarlos a los conocimientos científicos del momento y difundir adecuadamente sus logros en los foros sanitarios y sociales.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Caimari Jaume, María, Hospital Universitario Son Espases Baleares; Casano Sancho, Paula Hospital Sant Joan de Déu, Barcelona; Grau Bolado, Gema, Hospital Cruces, Vizcaya; Muñoz Calvo, M. Teresa, Hospital Infantil Niño Jesús, Madrid; Rial Rodríguez, José Manuel, Hospital Universitario Nuestra Señora de Candelaria, Santa Cruz de Tenerife, y Temboury Molina, Carmen, Hospital del Sureste, Madrid.
Los miembros del Grupo de Trabajo de Tiroides de la Sociedad Española de Endocrinología Pediátrica (SEEP) se presentan en anexo 1.
Para la realización de este trabajo se ha utilizado el instrumento AGREE: The AGREE Collaboration. AGREE Instrument Spanish versión (www.agreecollaboration.org).

Anales de Pediatría sigue las recomendaciones para la preparación, presentación y publicación de trabajos académicos en revistas biomédicas