
P158
LINFOMA DE CÉLULAS B EN PACIENTE DIAGNOSTICADO DE ATAXIA-TELANGIECTASIA
B. Nievas Soriano, C. Gil López, B. Losada Pinedo, S. Rueda Esteban, Jaime Campos Castelló, Francisco Valverde Moreno, Hospital Clínico Universitario San Carlos, Madrid.
Introducción: Se presenta el caso de un paciente de 10 años diagnosticado de ataxia-telangiectasia que desarrolló un linfoma de células B de alto grado.
Caso Clínico: Varón que presenta desde los 16 meses de vida marcha torpe. Desde los 4 años se asocian procesos infecciosos pulmonares recurrentes y telangiectasias oculares. De los antecedentes familiares destacaban la presencia de telangiectasias faciales en el padre y un carcinoma de útero materno. En el momento de consultar el paciente, a los 5 años, presenta carrera torpe sin ataxia, telangiectasias oculares y presencia de canas, así como babeo. La auscultación pulmonar mostraba estertores finos bilaterales.
En los estudios realizados, los datos más significativos consistieron en una alfa-fetoproteína de 102 UI/ ml, importante déficit de Ig A y linfopenia con escasa respuesta a mitógenos. En la RMN cerebral se evidenció una atrofia pancerebelosa. En el estudio genético se detectaron las mutaciones 8977C > T(ter) y 9177C > T.
Los cuadros respiratorios fueron aumentando en frecuencia y fue diagnosticado de bronquiectasias. Se añadió un cuadro de ataxia progresiva que terminó por imposibilitarle la deambulación a los 9 años, así como apraxia oculomotora, movimientos distónicos, disartria y temblor de intención.
Con esa edad presentó un cuadro de fiebre y aumento de adenopatías generalizadas. El aspirado de médula ósea fue morfológicamente normal. Presentaba una gammapatía monoclonal IgM-Kappa. El estudio A-P ganglionar mostró un linfoma linfoblástico de célula grande de tipo B.
Dado el estado general del paciente y su previsible evolución, los padres rechazan el tratamiento del proceso, y únicamente se administra tratamiento paliativo. Dos meses después el paciente fallece de una reagudización respiratoria.
Conclusiones: La ataxia-telangiectasia es un cuadro multisistémico con origen en la alteración del gen ATM, localizado en el cromosoma 11q22-23, y que interviene en la regulación del ciclo celular. Destacan datos como la evolución neurológica, el déficit de inmunidad con infecciones sinopulmonares, la sensibilidad a las radiaciones ionizantes y la propensión al desarrollo de procesos malignos. Por este motivo deben ser estudiados y seguidos de forma multidisciplinaria.
P159
TROMBOSIS DE CAVA Y AURÍCULA DERECHA EN TUMOR DE WILMS
J. Rodríguez Delgado, L. Martín Jiménez, J. Guerrero Fernández, A. Herrero Díez y P. García de Miguel
Hospital Universitario La Paz, Madrid.
Introducción: Presentamos un caso de tumor de Wilms con infiltración de vena cava y aurícula derecha.
Caso clínico: Niño de 8 años que comienza con astenia, anorexia y palidez. Una semana más tarde presenta dos episodios de hematuria macroscópica, por lo que se realiza una ecografía objetivándose una tumoración renal derecha. Antecedentes personales y familiares sin interés. Exploración física al ingreso: hígado a 1 cm. Abdomen distendido. Resto normal. Pruebas complementarias: Hemograma normal. Bioquímica: GOT 46 U/L. GPT 65 U/L. Resto normal. Coagulación normal. Ecografía abdominal: masa renal derecha 9,3x9,5x7,4 cm sólida y con áreas de hemorragia. Trombosis de la vena renal derecha, así como un gran trombo en la cava que se extiende desde la salida de las renales hasta la aurícula derecha. Riñón izquierdo normal. TAC toraco-abdominal: gran masa renal derecha ocupando los 2/3 inferiores del riñón, con invasión del espacio perirrenal y la grasa pararrenal posterior. Trombosis de vena renal derecha, cava y aurícula derecha con signos de neovascularización. Vena renal izquierda permeable. Ascitis. También se realizaron ecocardiograma y angio-resonancia torácica precirugía, confirmando el diagnóstico. Evolución: dada la extensión tumoral es ingresado en la UCIP durante el inicio de la quimioterapia precirugía, siendo intervenido un mes y medio más tarde, con extirpación completa de tumor y trombo, sin complicaciones. Diagnóstico anatomopatológico: tumor de Wilms.
Discusión y conclusiones: El tumor de Wilms es la neoplasia renal más común en la infancia. La trombosis de la cava es una complicación poco frecuente de este tumor (4-9%), siendo excepcional (0,7-1,3%) que el trombo llegue a afectar cavidades cardiacas. Habitualmente cursa de forma asintomática, por lo que debe buscarse siempre el trombo con ecografía en este tipo de tumores. El pronóstico en cuanto a supervivencia no depende de su extensión y es similar al de otros pacientes sin trombo con el mismo estadio y tipo histológico, siendo muy bajo el riesgo de embolismo. Esto permite el tratamiento habitual con quimioterapia preoperatoria para reducir el tamaño del tumor y del trombo, demorando la cirugía.
P160
SIGNOS OCULARES COMO PRIMERA SEÑAL DE ALARMA DE PROCESOS MALIGNOS INFANTILES
C. Calvo Monge, A. Fernández Landaluce, I. Astigarraga Aguirre, R. Martínez Fernández, A. Urberuaga Pascual, A. Fernández-Teijeiro Álvarez y A. Navajas Gutiérrez
Hospital de Cruces, Cruces-Barakaldo.
Diversas manifestaciones a nivel ocular pueden ser el primer signo de un tumor maligno en el niño. Se asocian tanto a procesos malignos localizados en el ojo o en la órbita como a tumores a distancia. Su reconocimiento es importante para un diagnóstico precoz del cáncer infantil, que permitiría una mejor respuesta al tratamiento y un mejor pronóstico, con disminución de las secuelas tanto a nivel ocular como general.
Presentamos una revisión de niños diagnosticados de tumores malignos, en los que la primera manifestación o el primer signo de alerta fue un problema a nivel ocular. Se analizan algunas malformaciones congénitas oculares como la aniridia y su asociación con el tumor de Wilms. Destacamos la leucocoria como manifestación típica del retinoblastoma, tanto en las formas bilaterales y familiares como en los casos esporádicos. La tumefacción palpebral y la proptosis o exoftalmos pueden ser debidos a procesos malignos como el rabdomiosarcoma orbitario o incluso metastásicos como el neuroblastoma. Algunas formas que parecen celulitis o uveítis pueden corresponder a cloromas o masas debidas a infiltración por células de leucemia. Diversos movimientos oculares anormales como el nistagmus o el flutter ocular pueden ser debidos a tumores cerebrales a nivel de cerebelo o en las vías ópticas y el opsoclonus puede asociarse a neuroblastomas a nivel abdominal o torácico. Muchos niños con tumores cerebrales presentan clínica de diplopia o afectación de pares craneales y el estudio del fondo de ojo con el edema de papila característico, permite sospechar la hipertensión endocraneal y el tumor cerebral.
Las manifestaciones oculares se asocian en muchos casos a otros síntomas o signos y por ello se debe realizar una historia clínica y una exploración física completa. El examen oftalmológico debe ser realizado por un especialista y el pediatra debe valorar conjuntamente las manifestaciones oculares con el resto de los problemas que puede presentar el niño. Diversas pruebas complementarias de imagen como TC y RM ayudan a localizar la lesión y se deberá realizar siempre una biopsia para confirmar el tipo de tumor. El tratamiento con cirugía, quimioterapia o radioterapia dependerá del tipo histológico pero deberá complementarse con una buena terapia ocular como la corrección de los déficits visuales o la colocación de prótesis oculares. El diagnóstico y tratamiento precoz de estos niños permitirá alcanzar una mejor supervivencia y una mayor calidad de vida.
P161
TUMOR RABDOIDE PRIMARIO: APORTACIÓN DE UN NUEVO CASO
L. Martín Jiménez, J.A. Ruiz Domínguez, A. Sastre Urgelles, S. Garcimartín Arévalo, M. Martínez Ruiz, S. Simo Segovia, I. Pastor y P. García de Miguel
Hospital Universitario La Paz, Madrid.
Introducción: Presentamos un caso de tumor rabdoide primario hepático (TRH) de frecuencia excepcional (11 casos publicados bien documentados).
Caso clínico: Niño de 8 meses con cuadro de fiebre sin foco de 15 días de evolución y hepatomegalia. Mediante ecografía y RM, realizada en otro centro, se confirma gran masa que infiltra todo el hígado con áreas quísticas focales. Marcadores tumorales (AFP, CEA y HCG) en rango normal. Con el diagnóstico de hemangioendotelioma se inicia tratamiento con corticoides. Reingresa al mes, por aumento de la masa, derrame pleural y ascitis. La angiografía previa a embolización descarta el diagnóstico inicial. Trasladado a nuestro hospital se realiza pleurocentesis, encontrándose células muy indiferenciadas y malignas en el líquido pleural. La biopsia fue diagnóstica. En el estudio de extensión con TC toracoabdominal se objetivan adenopatías mediastínicas. Se inicia tratamiento quimioterápico según protocolo SIOP MMT95, falleciendo el niño a los 2 meses por progresión tumoral.
Discusión: Neoplasia de frecuencia excepcional y origen incierto aunque parece tratarse de un tumor muy indiferenciado de posible origen neural. La edad media de presentación es de 9 meses. Cursa con fiebre sin foco y clínica secundaria a la masa abdominal.
El término rabdoide alude al parecido de las células con las de estirpe muscular. El tumor es infiltrante localmente con metástasis pulmonares y linfáticas. Son frecuentes los fenómenos de hemorragia y necrosis en el seno del tumor. Microscópicamente, son características las inclusiones citoplasmáticas para citoqueratinas y vimentina. El diagnóstico es anatomopatológico, debiendo obtenerse la muestra por biopsia y no por punción; son fundamentales los estudios inmunohistoquímicos y de microscopía electrónica para asegurar la fiabilidad del diagnóstico.
Conclusiones: Se debe incluir esta entidad en el diagnóstico diferencial de tumor hepático primario con AFP normal. Es importante la obtención de muestra suficiente de tejido por biopsia y la realización de estudios inmunohistoquímicos y de microscopía electrónica para asegurar un diagnóstico y establecer el pronóstico. El desenlace es fatal en todos los casos publicados.
P162
HEPATOBLASTOMA EN NIÑOS
M. González López, C. Vida Fernández, T.J. Martínez Arán, R. Maese Heredia, A. Herrero Hernández, M.J. Ortega-Acosta, T. Acha García y A. Jurado Ortiz
Hospital Materno Infantil, Málaga.
Introducción: El hepatoblastoma (HB) es el tumor hepático maligno más frecuente en niños. El tratamiento óptimo es quirúrgico, incrementándose el nº de tumores resecables desde la introducción de la quimioterapia (QT) previa. El pronóstico tras la resección completa es bastante bueno siendo la supervivencia más baja para los HB irresecables o recurrentes.
Material y métodos: Revisión retrospectiva de los 8 casos diagnosticados y tratados de HB (con confirmación histológica) en los últimos 15 años.
Resultados: Ver tabla a pie de página.
Conclusiones: Los HB presentan como clínica inicial más frecuente una masa abdominal, localizada en la mayoría en lóbulo derecho. Existen metástasis al diagnóstico en un 10-20% por lo que se debe realizar Rx y TAC de tórax. El aumento de la AFP es el test diagnóstico más característico, niveles normales se relacionan con la variante "anaplásica" y un pronóstico peor. El objetivo del tratamiento es conseguir una resección completa para lo cual la QT prequirúrgica incrementa el número de tumores resecables.

P163
QUISTE ÓSEO ANEURISMÁTICO DE MALA EVOLUCIÓN. REVISIÓN DE CASOS
R. Maese Heredia, M. González López, C. Vida Fernández, R. Vera Medialdea, T.J. Martínez Arán, A. Herrero Hernández y A. Jurado Ortiz
Hospital Materno Infantil, Málaga.
Introducción: El quiste óseo aneurismático (1% de los tumores óseos primarios) es una lesión lítica expansiva no neoplásica, generalmente metafisaria. Se da algo más en niñas en torno a los 13 años con formas primaria o secundaria a otras lesiones, sobre todo al tumor de células gigantes. Puede permanecer silente e incluso regresar o causar gran destrucción ósea. Son más frecuentes en los huesos largos, con un 12-30% en columna vertebral, mostrando imágenes radiológicas típicas.
Caso clínico: Varón de 12 años con cuadro de 3 días de evolución con impotencia funcional de MMII, dificultad para la marcha, caídas frecuentes, parestesias descendentes desde hipogastrio e hiperalgesia cutánea abdominal. A la exploración descenso de fuerza de MMII, más en MID, exaltación de ROT y sensibilidad conservada. Evoluciona a paraparesia extrema y afectación sensitiva progresiva. El LCR y el EMG no presentan datos patológicos. Se realiza RMN apreciándose lesión lítica vertebral a nivel de D5. Se practica exéresis parcial, laminectomía, siendo el informe AP correspondiente a quiste óseo aneurismático. Dada la no mejoría se completa la exéresis y se realiza artrodesis D4-D6. Desciende el nivel sensitivo y se recupera parcialmente la sensibilidad profunda, manteniéndose la paraplejia.
Revisión de casos: Desde 1995 se han diagnosticado en nuestro centro:

Conclusiones: Aunque en general se trata de un proceso benigno, puede ser agresivo localmente, siendo esencial identificar los casos secundarios para planificar una terapéutica correcta. El tratamiento de elección es quirúrgico, desde el curetaje a la resección amplia. El pronóstico libre de enfermedad suele ser excelente, siendo muy baja la tasa de recurrencias cuando se consigue una resección completa.
P164
INCIDENCIA DE TUMORES MALIGNOS INFANTILES EN LA COMUNIDAD DE CANTABRIA
M. San Román Muñoz, S. García Calatayud, M. Uyaguari Quezada y E. Pérez Gil
Hospital Universitario Marqués de Valdecilla, Santander.
Antecedentes: El Registro Nacional de Tumores Infantiles constituye la referencia epidemiológica del cáncer infantil en España. La comunidad de Cantabria nunca ha participado de forma completa en estos registros y tampoco se ha realizado ninguna casuística de cáncer infantil en esta comunidad.
Objetivos: Conocer la incidencia, distribución y epidemiología de los tumores malignos infantiles en Cantabria.
Material y métodos: Estudio retrospectivo realizado mediante revisión de las historias clínicas de 89 pacientes menores de 15 años con diagnóstico de tumor maligno en el período de 1995 a 2000, en el Hospital Marqués de Valdecilla. Las variables analizadas incluyen sexo, fecha de nacimiento, fecha de diagnóstico, localidad de residencia y diagnóstico histológico. Para el cálculo de incidencias se consideró el número de habitantes y su distribución por sexo y edades en esta comunidad, tomando como referencia el censo de Cantabria del año 1999. El análisis de los datos se realizó con los programas informáticos Microsoft Excel y SPSS v9.0.
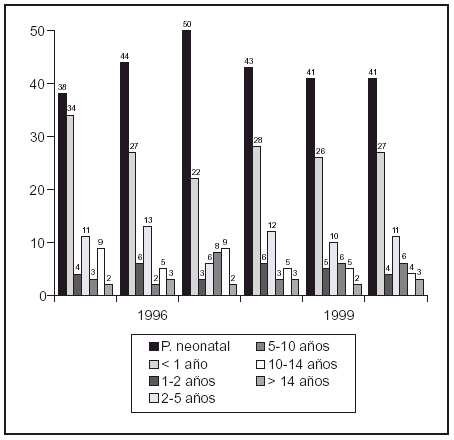
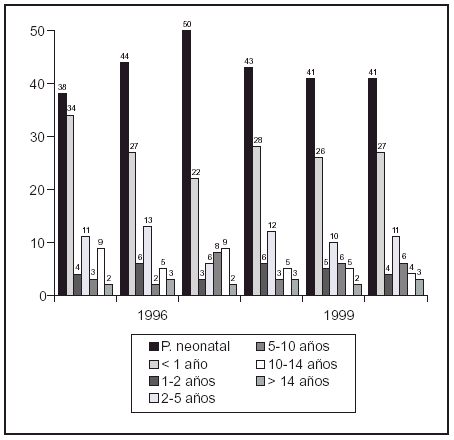
Resultados: La incidencia del cáncer infantil en Cantabria es de 153,5 casos nuevos al año por millón de habitantes menores de 15 años. El cáncer infantil predomina en niños residentes en zonas rurales, en menores de 5 años y en los varones. Los procesos malignos más frecuentes fueron las leucemias (32,6%), seguidas por los tumores del sistema nervioso central (23,6%) y del sistema nervioso simpático (11,2%). Comparando los sexos, en los varones predominan neuroblastomas y tumores del sistema nervioso central y en las mujeres, linfomas y tumor de Wilms. Respecto a la edad en los preescolares predominan las leucemias y neuroblastomas, en los escolares las leucemias y los tumores del sistema nervioso central y en los adolescentes aparecen los tumores óseos y linfomas.
Conclusiones: Las características epidemiológicas observadas en este estudio sobre el cáncer infantil en la comunidad de Cantabria no difiere en gran medida de los resultados obtenidos en estudios similares realizados en otras comunidades de España aunque llama la atención la alta incidencia de tumores del sistema nervioso simpático y la baja de linfomas.
P165
PRESENTACIÓN CLÍNICA Y RETRASO DIAGNÓSTICO DE 89 TUMORES MALIGNOS INFANTILES
E. Pérez Gil, S. García Calatayud, M. Uyaguari Quezada y M. San Román Muñoz
Hospital Universitario Marqués de Valdecilla, Santander.
Antecedentes: El cáncer infantil representa la segunda causa más frecuente de muerte en niños mayores de un año. Las mejoras terapéuticas y el diagnóstico precoz se relacionan con un aumento en su supervivencia. A menudo los síntomas y signos de presentación clínica no sugieren el diagnóstico de cáncer.
Objetivos: Conocer las características clínicas iniciales de los tumores malignos infantiles y su demora diagnóstica en la Comunidad de Cantabria.
Material y métodos: Estudio retrospectivo realizado mediante revisión de las historias clínicas de 89 pacientes menores de 15 años con diagnóstico de tumor maligno en el período de 1995 a 2000, en el Hospital Marqués de Valdecilla. Las variables analizadas incluyen síntomas y signos al diagnóstico, que se clasificaron en 3 categorías: altamente sugerentes, sugerentes y poco sugerentes de cáncer. Se recogió también el tiempo transcurrido desde el inicio de la clínica hasta el diagnóstico. El análisis de los datos se realizó con los programas informáticos Microsoft Excel y SPSS v9.0.
Resultados: De todos los datos clínicos recogidos, la fiebre-febrícula fue el signo más frecuente (29,2%), seguido de la astenia-anorexia (19,1%). El 76,1% de los síntomas referidos por los pacientes son clasificados como poco sugerentes de cáncer. El dato clínico sugerente de cáncer más frecuente fue la focalidad neurológica (13,5%), seguido del hallazgo de una masa no sugerente de adenopatía (10,1%). La demora diagnóstica media fue de 9,9 semanas para el conjunto de los tumores, observándose la mayor demora en los tumores del sistema nervioso central (11,3 semanas). Se describe una relación estadística directa entre el carácter inespecífico de los síntomas y signos de presentación y el tiempo de demora diagnóstica.
Conclusiones: El cáncer infantil debuta frecuentemente con signos y síntomas inespecíficos. Teniendo en cuenta, la relación estadística descrita entre una historia clínica inicial poco sugerente y el retraso en el diagnóstico de cáncer infantil, y que, el diagnóstico precoz mejora su pronóstico, el pediatra debe estar alerta ante síntomas frecuentes y comunes en la práctica diaria que pueden ser la forma de presentación de un cáncer infantil.
P166
TUMORES ÓSEOS MALIGNOS MÁS FRECUENTES EN LA INFANCIA
A.F. Bajo Delgado, M. Martínez Martínez, M. Díaz Suárez, A. Carboñé Bañeres y C. Calvo Escribano
Hospital Miguel Servet, Zaragoza.
Objetivo: Revisar la incidencia y supervivencia de los tumores óseos malignos (TOM) más frecuentes en la infancia, Sarcoma de Ewing (SE) y Osteosarcoma (Os) en nuestro hospital.
Material y métodos: Estudio retrospectivo epidemiológico, de los 26 pacientes afectos de TOM (Os. y SE) tratados en nuestro hospital desde 1981-2001. Todos los pacientes fueron tratados de acuerdo a los protocolos nacionales vigentes en cada época. El cálculo de la supervivencia global (SG) y supervivencia libre de eventos (SLE), se realizo por el método actuarial de Kaplan y Meyer.
Resultados: Incidencia de TOM: 4%. De los 17 casos de SE, 35% son niñas y 65% niños. Edad media de presentación: 9 años y 6 meses (rango: 1-17 años). Localización: Axial: 6 casos; extremidades inferiores (EI) 6; 3 costales; 1 craneal; 1 en clavícula. Motivo de consulta más frecuente: tumefacción y dolor de 9 semanas de evolución como media. Metástasis al diagnóstico: 5 casos (4 en pulmón, 1 ósea). Componente histológico: Ewing típico: 12 casos; Tumor maligno de célula redonda pequeña sugerente de Ewing: 5. SG a 5 años 42%, SLE: 32% de toda la serie. En los casos diagnosticados entre 1981-1990: SG: 28% y entre 1990-2001 la SG es del 77% y 58% a los 3 y 5 años respectivamente y una SLE de 33% a 5 años.
De los 9 casos de Os, 25% niñas y 75% niños. Edad media de presentación: 11 años y 3 meses (rango: 6-17 años). Localización: Axial 1 caso; de EI: 8. Motivo de consulta más frecuente: dolor e impotencia funcional con una media de 8 semanas de evolución. Metástasis al diagnóstico en 1 caso (localización ósea). Componente histológico: 5 condroblásticos, 3 osteoblásticos, en 1 no consta. Dado la escasa muestra y la amplitud del tiempo considerado, sólo se realiza el análisis de SG a 5 años, de toda la serie: 50%.
Conclusiones: Mayor incidencia de TOM en niños, localización predominante: EI. Clínica más frecuente: dolor e impotencia funcional de 2-3 meses de evolución. Edad de presentación: mayor en Os (11 años) que en SE (9 años). La supervivencia es baja, a pesar de haber mejorado en la última década.
P167
ENTEROCOLITIS EN NIÑOS NEUTROPÉNICOS
C. Montero Valladares, G. Ramírez Villar, C. Márquez Vega, C. Muñoz Román, G. Calderón López, M.L. Anguita Quesada, A.M. Álvarez Silván, M.S. Camacho Lovillo, A. Valladares Otero y A. Vilaplana López
Hospital Universitario Virgen del Rocío, Sevilla.
Antecedentes y objetivos: La enterocolitis neutropénica (ECN) o tiflitis es un proceso inflamatorio que afecta a la mucosa intestinal y que se desarrolla en pacientes neutropénicos debido a quimioterapia intensiva por procesos malignos. Nuestro objetivo ha sido valorar la relación de esta patología con distintos tipos de malignancias y tratamientos citostáticos, así como su presentación clínica, tratamiento y evolución.
Métodos: Se han revisado los niños menores de 15 años con tumores sólidos y linfomas tratados en el servicio de Oncología del Hospital Infantil Virgen del Rocío durante los años 2000-2001.
Resultados: De todos los pacientes revisados hemos encontrado un total de 5 casos, con una edad media de 10 años. El diagnóstico previo fue de linfoma en 2 casos y de tumores sólidos en 3 casos (rabdomiosarcoma, sarcoma de Ewing y tumor germinal maligno). Los citostáticos más usados previamente al desarrollo de la enfermedad fueron vincristina y adriamicina. La presentación clínica más frecuente consistió en afectación del estado general, fiebre, neutropenia intensa, dolor y distensión abdominal, vómitos y deposiciones diarreicas; sólo en 2 casos se evidenció una hemorragia digestiva. El traslado a UCI fue preciso en 3 de nuestros pacientes. En las pruebas de imagen se evidenció un edema de la pared del intestino y la presencia de ascitis. Sólo en 1 paciente el hemocultivo fue positivo a Enterococo. En cuanto al tratamiento, todos los pacientes precisaron reposo digestivo y nutrición parenteral, así como antibióticos de amplio espectro, inotrópicos, transfusión de hemoderivados, diuréticos, seroalbúmina, etc. La duración media del cuadro fue de 20 días y la mortalidad fue de un 20% (1/5).
Conclusiones: La ECN tiene una alta morbimortalidad, por tanto es muy importante la sospecha diagnóstica y la instauración precoz del tratamiento. Se trata de una enfermedad en la que debemos pensar como complicación de todo paciente oncológico con fiebre neutropénica, afectación del estado general, dolor y distensión abdominal.
P168
RESTOS NEFROGÉNICOS EN LA ANATOMÍA PATOLÓGICA DE TRES CASOS DE SOSPECHA DE WILMS BILATERAL
A. de Andrés González, G. Lunar Soriano, N. Martín Ramos, M.A. Díaz, C. López Vilar y L. Madero Rodríguez
Hospital Universitario Marqués de Valdecilla, Santander.
Introducción: El nefrobastoma o tumor de Wilms es el tumor renal maligno más frecuente en la infancia. Tumor embrionario, capaz de metastatizar a distancia, muy sensible a radioterapia y quimioterapia, curable en un 85% de los casos. Un 4-6% son bilaterales y sincrónicos.
Casos clínicos: Presentamos tres casos de sospecha por técnicas de imagen de tumor de Wilms bilateral.
Caso 1: Niña de 2 años y 6 meses con fiebre y síntomas catarrales en cuya exploración física se palpa masa abdominal sólida desde hipocondrio izquierdo hasta fosa ilíaca izquierda. Ecografía y TAC abdominal: masa sólida en hemiabdomen izquierdo dependiente del riñón y dos lesiones en riñón derecho. Se comienza tratamiento quimioterápico prequirúrgico. Tras cirugía la anatomía patológica (AP) revela nefroblastoma estromal estadio II izquierdo y restos nefrogénicos derechos. Actualmente en tratamiento quimioterápico poscirugía.
Caso 2: Niño de 8 años y 6 meses con astenia palidez y tumoración en hipocondrio izquierdo a la palpación. Ecografía y TAC abdominal: sospecha de nefroblastoma bilateral. AP tras quimioterapia: nefroblastoma estándar izquierdo, nefroblastoma nodulo-epitelial con áreas de células blastomatosas perilobares.
Caso 3: Lactante de 6 meses con masa abdominal, tras cirugía se confirma en AP la existencia de tumor de Wilms bilateral. Controles posteriores tras quimioterapia localizan dos nuevos nódulos en riñón derecho que tras biopsia son diagnosticados de restos nefrogénicos.
Conclusiones: Los restos nefrogénicos son considerados lesiones precursoras de tumor de Wilms, constituidos por pequeñas o microscópicas agrupaciones de células blastomatosas, con frecuencia multicéntricos. Ante la sospecha de tumor de Wilms bilateral, tener presente la posibilidad de la existencia de restos nefrogénicos. Una vez confirmada esta existencia se deberán hacer controles seriados de ambos riñones durante largo tiempo, ante la posibilidad de la aparición de tumor de Wilms metacrómico.
P169
LEIOMIOSARCOMA EN POST-TRASPLANTE HEPÁTICO. A PROPÓSITO DE UN CASO
J. Fernández Fernández, L. Martín Jiménez, M.C. Díaz Fernández, M.C. González Martín, A. Herrero Díez y P. García de Miguel
Hospital Universitario La Paz, Madrid.
Introducción: Presentamos un paciente pediátrico, postrasplantado hepático, en tratamiento inmunosupresor y serología positiva para virus de Epstein-Barr (VEB), en el que se diagnostica un leiomiosarcoma a los 3 años del trasplante.
Caso clínico: Niña de 3 años y 9 meses, diagnosticada al mes y medio de vida de atresia de vías biliares extrahepáticas. Recibió trasplante hepático a los 6 meses de vida, siguiendo tratamiento inmunosupresor con ciclosporina y prednisolona. No presentó complicaciones significativas postrasplante salvo hemorragias digestivas secundarias a trombosis portal. Serología positiva para VEB(viremia negativa desde hacía un año). En control ecográfico abdominal, realizado a los tres años postrasplante, se observa masa entre las asas intestinales de localización supravesical. La TAC y RMN abdominal objetiva una lesión focal en situación anterior al polo inferior hepático. Así mismo se evidencia otra lesión focal, de aproximadamente 1 cm adyacente a la porta sin invadirla. No se observan adenopatías retroperitoneales. En la RX de tórax se halla un aumento del hilio derecho e infiltrados pulmonares, confirmándose en el TAC torácico como imágenes sugestivas de metástasis. Se realiza biopsia guiada de la lesión principal, observándose en el examen ultraestructural la presencia de rasgos de diferenciación muscular lisa en las células tumorales. Con técnicas de PCR se obtuvo intensa positividad frente al EBNA2 y LMP1 del VEB. El diagnóstico anatomopatológico fue de Leiomiosarcoma. En la actualidad la paciente recibe tratamiento quimioterápico según el protocolo de la Sociedad Española de Pediatría.
Discusión: 1) El leiomiosarcoma es una neoplasia mesenquimal maligna extremadamente rara en niños. 2) Los casos hallados tienen en común el haber recibido un trasplante de órgano sólido en un período previo de 2 a 4 años, tratamiento inmunosupresor y serología positiva para VEB. 3) Los pacientes inmunosuprimidos presentan alto riesgo de desarrollo de neoplasias. 4) La incidencia de positividad para VEB es mayor en pacientes inmunocomprometidos. los mecanismos 5) La infección latente por VEB, sería la responsable de los trastornos linproliferativos que, sobre la base de inmunosupresión de estos pacientes, precipitarían los mecanismos carcinogénicos.
