
P150
SÍNDROME UROFACIAL DE OCHOA
J.A. López Medina, P. Lupiani Castellanos, B. Bravo Mancheño, D. Barajas de Frutos, J. Pedrero Vera, A. Vicente Pintor, J. Pacheco Sánchez, R. López-Jurado Romero de la Cruz, J.L. Barrionuevo Porras y L. Ortega Martos
Hospital Universitario Virgen de las Nieves, Granada.
Antecedentes: El síndrome urofacial o de Ochoa consiste en la asociación de una facies típica de inversión de la expresión facial al sonreír (al invertirse la boca, la sonrisa se asemeja al llanto), con una disfunción vesical no neurogénica, ni debida a obstrucción orgánica. Si la disfunción vesical pasa desapercibida o no es tratada, secundariamente, puede aparecer afectación del tracto urinario superior en forma de dilataciones, nefropatía de reflujo (RVU) o incluso insuficiencia renal.
Caso clínico: En el año 1996, nuestra paciente fue hospitalizada con un mes de vida, por una pielonefritis aguda. En la exploración física destacaba una facies dismórfica con epicantus y filtrum alargado. Tenía antecedentes familiares de nefrouropatía. Entre las pruebas complementarias iniciales, el urocultivo fue positivo a Escherichia coli, la función renal fue normal, encontrándose un aumento de la β2microglobulina en orina (1.591 mg/l) . Cariotipo normal. Ecografía renal: ureterohidronefrosis bilateral. Cistografía (CUMS): RVU bilateral, derecho grado IV, izquierdo grado V e intrarrenal. Gammagrafía renal (DMSA): hipocaptación generalizada del riñón izquierdo (RD 66%, RI 34%). A pesar de la quimioprofilaxis presentó infecciones urinarias (ITUs) de repetición, motivo por el cual se realizó reimplantación ureteral tipo Cohen, al año de edad, El reflujo se resolvió, sin embargo, continuó con ITUs de repetición hasta los tres años. Alrededor de esta fecha, se detectó la facies típica de inversión facial al sonreír, lo que indujo a investigar una disfunción vesical subyacente (clínicamente no evidente). El estudio urodinámico reveló una vejiga inestable, de altas presiones y baja acomodación. Actualmente, el tratamiento está dirigido a reducir las presiones intravesicales en forma de anticolinérgicos y vaciamiento vesical completo.
Conclusión: La expresión facial de estos enfermos permite la deteción precoz de una disfunción vesical, que probablemente esté en el origen de las uropatías asociadas, y prevenir o paliar el daño renal. Conseguir reducir las presiones vesicales, con ejercicios de vaciamiento, medicación o cateterismo intermitente, así como prevenir las infecciones urinarias constituyen los pilares del tratamiento. Los antecedentes familiares cargados de uropatía en esta paciente sugieren la existencia de formas incompletas del síndrome de Ochoa que no asocien la inversión facial.
P151
LITIASIS RENAL INFANTIL: ANÁLISIS DE 56 PACIENTES
A. de Andrés, G.A. Martos, M. Cortés, J.L. Ecija y M. Vázquez Martul
Hospital del Niño Jesús, Madrid, Universidad Autónoma de Madrid, Madrid.
Objetivo: Aportar y analizar los hallazgos clínicos, etiológicos y factores favorecedores e inhibidores de la litogénesis en la edad pediátrica.
Material y métodos: Se estudian en una sección de nefrología pediátrica 56 pacientes, 34 niños (60%) y 22 niñas (39,3%) con edades comprendidas entre 1 mes y 17,8 años (media 6,37). El período de seguimiento osciló entre 1 y 15,2 años (media 5,3). A todos los pacientes se les realizó un estudio de función y morfología renal así como determinación de la excreción urinaria de ácido úrico, calcio, magnesio, citrato y aminoácidos
Resultados: Presentaron clínica de dolor el 55,3% de los pacientes, hematuria el 46,4%, síntomas miccionales el 21,4%. Como asociaciones más frecuentes encontramos hematuria + dolor en el 18% y hematuria + dolor + síntomas miccionales en el 11%. El análisis fisico-químico del cálculo fue posible en 25 casos (46,3%) siendo entre ellos un 32% de oxalato cálcico, un 8% de fosfato cálcico, un 12% de fosfato amónico magnésico, un 8% de cistina, un 4% de ácido úrico y un 36% mixtos. De los 56 pacientes la etiología de la litiasis fue metabólica en el 42,8%, infecciosa en el 3,6% e idiopática en el 53,6%. Entre las causas metabólicas se encontraron la hipercalciuria en un 50%, hipomagnesiuria en un 4,5%, hiperoxaliuria en un 11,4%, cistinuria 7,7%, hiperuricosuria 7,7% e hipocitraturia 3,8%. Ningún caso presentó alteraciones del metabolismo ácido-base. La recidiva de la litiasis se encontró en 4 pacientes (7,2%) mientras que 47 pacientes (83,9%) no presentaban litiasis y en un 8,9% no se observó modificación de la misma al final de la evolución. Se halló familiaridad en 26 pacientes (46,4%). La sensibilidad diagnóstica de la ecografía abdominal fue del 87,4% y la de la radiografía simple de abdomen fue del 71%, no siendo significativa esta diferencia. La actitud terapéutica seguida fue conservadora en el 76,4% de los pacientes, quirúrgica en el 16,4% y litotricia en el 5,5%
Conclusiones: Predominio del sexo masculino. La hipocitraturia como causa etiológica metabólica ha sido muy rara, siendo la hipercalciuria la más frecuente. Escasas recidivas. Familiaridad en elevado porcentaje. Buen pronóstico de la litiasis renal en esta serie.
P152
ANOMALÍAS CARDÍACAS Y DIGESTIVAS ASOCIADAS AL SÍNDROME NEFRÓTICO CONGÉNITO TIPO FINLANDÉS
B. Orive Olóndriz, I. Hualde Tapia, L. Pérez Díaz y A. Rodríguez Estévez
Hospital Txagorritxu, Vitoria.
El síndrome nefrótico congénito tipo "Finlandés" es una rara enfermedad autosómica recesiva causada por mutaciones en el gen de la nefrina, NPHS1. La mayoría de los niños son prematuros, presentan proteinuria masiva de inicio intraútero y evolucionan hacia la insuficiencia renal crónica. La histología renal demuestra: dilatación quística de los túbulos proximales, hipercelularidad mesangial y fibrosis glomerular. Su asociación con anomalías cardiacas y digestivas se ha referido escasamente.
Objetivo: Analizar dos pacientes con SNC "finlandés" asociado con estenosis pulmonar y reflujo gastroesofágico. Estudio descriptivo:
Caso 1: Mujer de 37 semanas de gestación (septiembre 94). Pn: 2.490 gr, sin antecedentes familiares de síndrome nefrótico, diagnosticada en la primera semana de vida por edemas y proteinuria, fenotipo normal, cariotipo 46 XX. Biopsia renal: dilatación quística de túbulos contorneados proximales. Recibió tratamiento con Enalapril, L-Tiroxina, y eventuales perfusiones de albúmina. Se detectó un soplo sistólico 2/6 en foco pulmonar, apreciándose en ecocardiograma estenosis pulmonar supravalvular con gradiente de 40 mmHg. La cardiopatía permaneció estable. Diagnosticada de reflujo gastroesofágico, precisó tratamiento quirúrgico por mala respuesta al tratamiento médico. La función renal se deterioró progresivamente y a los 3,5 años recibió un transplante renal, falleciendo por rechazo agudo del injerto.
Caso 2: Mujer de 37 semanas de gestación (enero 99) Pn 1890 gr. Peso placenta: 900 gr (47% peso natal) diagnosticada al nacimiento por proteinuria masiva y edemas. Fenotipo normal, cariotipo 46XX, Biopsia renal: dilatación quística de túbulos contorneados proximales. Tratamiento: Enalapril, L-Tiroxina, y cisaprida. Se apreció soplo sistólico 2/6 en foco pulmonar, presentando en la ecocardiografía estenosis pulmonar supravalvular con gradiente de 17 mmHg, que permanece estable. Se diagnosticó reflujo gastoesofágico por phmetria, con buena evolución clínica. Actualmente en tratamiento conservador por insuficiencia renal crónica severa
Conclusiones: Es importante la valoración cardiológica para detectar alteraciones suceptibles de cirugía y prevención de endocarditis bacteriana. Diagnosticar y controlar el reflujo gastroesofágico patológico es fundamental para conseguir un adecuado estado nutricional
P153
HIPONATREMIA POR PÉRDIDA SALINA DE ORIGEN RENAL EN PACIENTE CON DIABETES INSÍPIDA CENTRAL
P. Betrián Blasco, R. Cabrerizo de Diago, M. Cuadrado Martín, B. Alonso del Val, E. García Jaria, D. Gros Estebán, M. Justa Roldán y C. Loris Pablo
Hospital Miguel Servet, Zaragoza.
Introducción: En la diabetes insípida central la aparición de hiponatremia puede deberse a exceso de dosis con ADH. Puede deberse también a un cuadro de pérdida salina. El síndrome cerebral pierde sal se ha descrito a menudo asociado a daño intracraneal. El cuadro se ha definido por pérdida renal de sodio en presencia de hiponatremia con depleción del espacio extravascular. Presentamos el caso de un paciente afecto de diabetes insípida central en el que se produjo un cuadro pierde sal.
Caso clínico: Paciente de 18 meses de edad diagnosticado de diabetes insípida central en que inicia tratamiento con Desmopresina con dosis de 10 µg/día. A los 2 meses de inicio de tratamiento presenta un cuadro de deshidratación hiponatrémica (Na: 108) con convulsiones generalizadas precisando ingreso en UCI. Se aprecia aumento de la natriuresis y poliuria requiriendo mayores dosis de ADH. Presenta posteriormente otros 2 episodios de deshidratación hiponatrémica. Se inicia aporte de suplemento oral de ClNa (34 mEq/día). Posteriormente, estando asintomático, se ha podido completar el estudio con determinación de péptido natriurético atrial siendo su valor normal. Actualmente ha sido retirado el tratamiento permaneciendo asintomático.

Comentarios: No se ha podido detectar la causa que desencadenó el cuadro de pérdida de sal de origen renal en el paciente, no objetivándose antecedente de daño intracraneal ni alteraciones en la RMN. Podría deberse a alguna alteración transitoria en el eje ADH y de péptidos natriuréticos que posteriormente se hubiera normalizado.
P154
TRATAMIENTO CONSERVADOR DEL REFLUJO VESICOURETERAL DE ALTO GRADO DIAGNOSTICADO EN PERÍODO NEONATAL
A. Rodríguez do Forno, C. Lorenzo-Legerén, G. Nóvoa Gómez, S. Rey García y A.I. Villares Porto-Domínguez
Hospital Cristal Piñor, Ourense.
Antecedentes: Las técnicas de imagen evidencian reflujo vesicoureteral (RVU) con frecuencia en el periodo neonatal. Es controvertida la actitud con el RVU de alto grado.
Objetivo: Comprobar la evolución con tratamiento conservador de niños diagnosticados en el 1º mes de vida de RVU primario. Es parte del estudio del Grupo Colaborativo Gallego de RVU Congénito.
Métodos: Pacientes con RVU grado III o superior (n = 42). 13 niñas (31%) y 29 niños (69%). Diagnostico secundario a anomalías ecográficas en 24 (64,3%) y precedido por una infección de orina (ITU) en 15 (35,7%). Excluimos los pacientes con patología obstructiva y/o anomalías asociadas en el riñón refluyente. Tiempo medio de seguimiento: 1,9 años (1a 4,7). Análisis descriptivo habitual. P. exacta de Fisher para comparación de v. cualitativas, Kaplan-Meier, prueba de log-rank. SPSS 10.0.
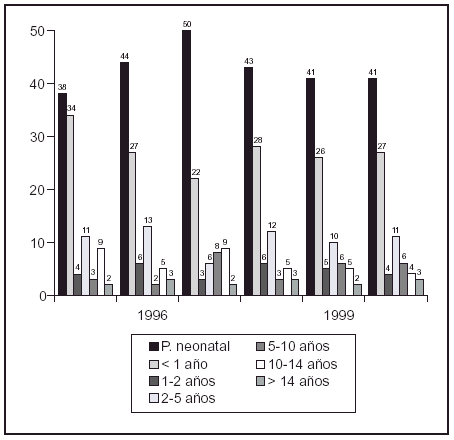
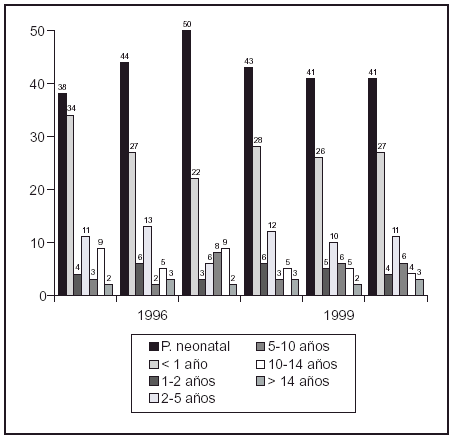
Resultados: Localización derecha 14 (33,3%), izquierda 10 (23,8%) y bilateral en 18 (42,9%). Número total de unidades refluyentes (UR) fue de 60. Grado III en 48 (80%), IV en 10 (16,6%), y V en 2 (3,3. El DMSA inicial fue anormal en 11/ 60 UR (18,3%) con displasia del riñón afectado en 5 (8,3%). Según sexo, el daño renal se produjo solo en los varones 8/29 (27,6%). Del total de UR se resolvió el RVU en 42/60 (70%). De los 48 de grado III, desapareció en 38 (79,2%); de los 10 de grado IV en 4 (40%), no resolviéndose en ninguno de los 2 de grado V. Mediana de tiempo hasta resolución espontánea de 20 meses; variando según grado de RVU, log-rank = 4,47, p = 0,03. Situación actual de 18 UR que no curan: de 10 con RVU inicial grado III, 7 son de bajo grado (I-II) y 3 UR están pendientes del primer control de cistografía. De los 6 grado IV, 3 son de grado III y 3 de grado II. Uno de grado V es de grado IV y en el otro se realizó cirugía.
Conclusiones: El tratamiento conservador del RVU congénito de alto grado es una opción adecuada. La resolución es significativamente menor en los grados IV y V. El daño renal se produjo sólo en los varones.
P155
NEFROCALCINOSIS: REVISIÓN DE 28 CASOS EN EDAD INFANTIL
B. Alonso del Val, E. García Jaria, D. Gros Estebán, L. Palacín González, M. Justa Roldán y C. Loris Pablo
Hospital Miguel Servet, Zaragoza, Hospital Clínico Universitario Lozano Blesa, Zaragoza.
Antecedentes: La nefrocalcinosis no es una entidad clínica única, sino una consecuencia de diferentes patologías. Consiste en depósitos difusos de sales de calcio, fosfato u oxalato en el parénquima renal. Entre las causas más frecuentes destacan: hipercalciuria idiopática, tubulopatías y la secundaria al uso de furosemida (sobre todo en periodo neonatal). Tanto la clínica como el tratamiento dependen de la enfermedad de base. El diagnóstico es fundamentalmente ecográfico.
Métodos: Se ha realizado estudio retrospectivo de los 28 casos diagnosticados en esta unidad hasta el momento actual excluyendo el periodo neonatal. Se ha analizando la etiología de las mismas y la posible evolución a insuficiencia renal crónica (IRC) .
Resultados: De los 28 casos revisados se han encontrado:

Conclusiones: Entre nuestra casuística predomina la nefrocalcinosis secundaria a tubulopatías, destacando en frecuencia el síndrome de hipercalciuria-hipomagnesemia. Son precisos estudios ecográficos y controles seriados de la función renal para detectar precozmente la posible evolución hacia insuficiencia renal.
P156
LITIASIS VESICAL ENDÉMICA, UNA PATOLOGÍA DEL SUBDESARROLLO
R. Calvo, J. Gutiérrez Blasco, D. Segura, O. García Bodega, M. Justa Roldán y C. Loris Pablo
Hospital Miguel Servet, Zaragoza.
Introducción: La litiasis vesical endémica presenta una clara distribución geográfica que incluye sudeste de Rusia, Balcanes, países del Magreb y Pérsicos, Israel y sur de China. La incidencia en estos lugares es de 10-100 veces mayor que en las zonas no endémicas.
En la patogenia parece intervenir una dieta pobre en proteínas animales, cereales y leche, rica en oxalatos, mala hidratación, junto con climas calurosos y secos. El principal constituyente de los cálculos es el oxalato cálcico. Presentamos dos casos recientes diagnosticados en nuestro hospital.
Caso 1: Niño de 4 años de edad, procedente de Gambia, llegado a nuestro país hace dos meses. Desde hace dos años presenta episodios de disuria, en ocasiones acompañados de fiebre.
Exploración física: Peso 12 Kg (< P3). Talla 104 cm (P25-50). Aspecto desnutrido. Exploraciones complementarias: Ac. úrico: 2,17 mg/dl. Fe: 48 mg/dl; prealbúmina 15,4 (18-30); RBP 1,87 (3-6) mg/dl. Función renal: GRF: 100 ml/mn/1,73 m2. TRP: 92,94%. Calciuria: 8 mg/Kg/24 h. Citraturia: 393 mg/l. Oxaluria: 10,2 mg/24 h. Aminoaciduria: Normal. Orina: Leucocituria. Urocultivo: Enterococo. Rx abdomen: Imagen radioopaca 2 x 2 cm. en vejiga. Eco renal: Litiasis vesical, resto normal. CUMS: No reflujo. Tratamiento: Cistotomía para extracción del cálculo. Evolución: favorable, sin reaparición de sintomatología.
Caso 2: Niño de 12 años. Participa en un programa de acogida de verano a niños saharauís. En la revisión rutinaria que se realiza en nuestro hospital la familia refiere polaquiuria. Exploración física: Sin hallazgos patológicos; peso 32 Kg (P10), talla 130 cm (< P3). Exploraciones cmplementarias: BQ: Normal. Función renal: GRF: 117,9 ml/mn/1,73 m2. Citraturia: 270 mg/l. Oxaluria: 18,3 mg/24 h. Aminoaciduria: Normal. Sedimento orina: Piuria. Urocultivo: negativo. Edad ósea: 9 años 6 meses. Rx de abdomen: Litiasis vesical 2 x 3 cm. UIV: Riñones y vejiga normales, sin fallo en vaciamiento; litiasis vesical de 2 cm de bordes espiculados. Análisis del cálculo: Oxalato cálcico dihidrato. Tratamiento: Cistotomía para extracción del cálculo. Evolución: favorable, sin reaparición de sintomatología.
Conclusiones: La litiasis vesical poco habitual en nuestro medio, aumenta su incidencia como consecuencia del fenómeno de la inmigración. La sospecha clínica en niños procedentes de las regiones endémicas, encamina el diagnóstico, fácil y sencillo. El tratamiento resulta rápido y eficaz. La evolución favorable si se corrigen las circunstancias socioeconómicas que la favorecen.
P157
POLIQUISTOSIS RENAL DOMINANTE Y ESCLEROSIS TUBEROSA: ENFERMEDAD DE GENES CONTIGUOS
V. Lanzadera Arencibia, M. Vázquez Martul y G.G. Mardones
Hospital del Niño Jesús, Madrid.
Antecedentes y objetivo: Se presenta una asociación infrecuente de dos enfermedades, poliquistosis renal dominante (PKD) y esclerosis tuberosa (TSC), que plantean un diagnóstico diferencial dentro de la clasificación de quistes renales. Existe ligamiento genético en el cromosoma 16 entre estas dos entidades, por lo que grandes delecciones en la zona que incluye ambos genes, resultan en la asociación de ambas.
Métodos: Se hace un análisis inicial y evolutivo de dos varones de 9 meses y 6 años de edad, con la mencionada asociación. Se han realizado estudios de función renal, exploraciones neurológicas e investigaciones por imagen renales y cerebrales, durante evoluciones de 15 y 3 años respectivamente.
Resultados:Caso 1. -Varón diagnosticado a los 9 meses de PKD, con abdomen globuloso, hipertensión arterial y nefromegalia y quistes renales bilaterales en ecografía renal. Diagnosticado a los 13 meses de TSC, con crisis convulsivas parciales, manchas acrómicas diseminadas y calcificaciones subependimarias y hamartomas tuberosos en TAC craneal. En su evolución presenta retraso mental, angiofibromas faciales, rabdomioma cardiaco y disminución progresiva de la función renal, con insuficiencia renal crónica moderada a los 13 años.
Caso 2. -Varón de 6 años de edad diagnosticado inicialmente de TSC, con crisis convulsivas, angiofibromas faciales y nódulos periventriculares y tuberomas corticales en TAC craneal, y posteriormente de PKD, con nefromegalia radiológica, hipertensión arterial, varias infecciones urinarias y grandes quistes renales bilaterales en ecografía renal. En su evolución presenta un astrocitoma interventricular. Durante los 3 años de seguimiento, se conserva la función renal. En ninguno de los dos casos existen antecedentes familiares de PKD.
Conclusiones: En el diagnóstico de PKD sin antecedentes familiares es obligado descartar TSC. Asimismo, en TSC, deberá realizarse precozmente ecografía renal y controles periódicos de la tensión arterial, por su posible asociación con PKD.
