




Introducción
El neuroblastoma es el tumor sólido extracraneal más frecuente de la infancia y se caracteriza por una evolución clínica heterogénea que abarca desde una progresión maligna rápida a una regresión espontánea. Como resultado, tanto el pronóstico como la respuesta al tratamiento varían ampliamente 1,2. La identificación de los factores pronósticos requiere siempre realizar una biopsia, y que esta sea estudiada en un laboratorio de referencia o con amplia experiencia. Existe una estandarización de criterios clínicos para diagnóstico, estadiaje, y respuesta al tratamiento conocida como International Neuroblastoma Staging System (INSS) 3, que se usan junto con datos biológicos (histopatológicos y genéticos) para crear nuevos grupos terapéuticos. Los análisis histopatológicos tienen un importante valor para conocer la biología tumoral 4,5. En 1984, Shimada et al6 describieron un sistema de clasificación basado en la cantidad de la estroma schwanniana, el grado de diferenciación, el índice de mitosis-cariorrexis (MKI) y la edad al diagnóstico, que distinguía dos grupos de pronóstico histológico: favorable y desfavorable . Actualmente se utiliza la International Neuroblastoma Pathology Classification (INPC) 7, basada en la anterior. Las anomalías genéticas características de estos tumores se conocen desde hace tiempo y algunas de ellas poseen claro valor pronóstico 8-10. Entre los marcadores genéticos de neuroblastomas agresivos se incluyen la diploidía o tetraploidía 11, la amplificación del gen MYCN (MNA) 12,13, la deleción (del) 1p36 14,15, y la ganancia de 17q (+17q) 16,17. Esta última reestructuración, que se presenta con una alta frecuencia, puede resultar de una translocación no balanceada del 17q con más de 20 regiones cromosómicas diferentes 18. Recientemente, se ha encontrado que los tumores con disomía del cromosoma 17 y un presumible estadio favorable pueden representar un subgrupo de riesgo alto 19. Además, se han descrito como marcadores de posible valor pronóstico las pérdidas frecuentes de heterozigosidad (LOH) en otras regiones cromosómicas, especialmente 2q (30 %), 3p (15,3 %), 4p (19,5 %), 9p (36 %), 11q (5-44 %), 14q (18-23 %) y 18q (31 %) 20-23. Se han descrito asociaciones entre pérdidas de 11q, 3p y 14q, que se correlacionan inversamente con MNA y del 1p, y que identifica subgrupos diferentes de tumores avanzados 20-22.
La edad, el estadio y MNA son los marcadores con valor pronóstico independiente. A ellos se añaden, con menor valor, la clasificación histológica de Shimada, la del 1p y la ploidía. Otros marcadores biológicos han demostrado valor pronóstico en estudios aislados, pero no han sido utilizados todavía en grandes estudios cooperativos para conocer cuál es su valor pronóstico real 24-26. Los enfermos se clasifican, por tanto, en grupos de riesgo con recomendaciones terapéuticas diferentes para cada uno de ellos. El grupo de alto riesgo (NB-HR) es el más numeroso (45 %), compuesto por pacientes mayores de un año con estadio 4, aunque también incluye a cualquier otro estadio de la enfermedad con MNA, excluyendo el estadio 1 27. El pronóstico de estos enfermos es malo, incluso con tratamientos muy agresivos. Sólo la MNA confiere un valor pronóstico negativo a los tumores que la presentan 28,29.
En este estudio se plantea la utilidad de la técnica de hibridación genómica comparada (CGH) que permite examinar el perfil genómico de un grupo de pacientes con pronóstico desfavorable para identificar anomalías cromosómicas adicionales que puedan ser útiles para la estratificación terapéutica.
Material y métodos
Pacientes
El material tumoral de los pacientes estudiados fue remitido entre enero 2002 a febrero 2005 al Centro de Referencia Nacional de Estudios Anatomopatológicos y Biológicos (Departamento de Patología, Universidad de Valencia) con el correspondiente consentimiento informado. La edad de los pacientes oscilaba entre 14 meses y 15 años. El diagnóstico de neuroblastoma y estadiaje fue realizado según la INSS: estadio 2/3 (n = 9), estadio 4 (n = 51). Fueron estudiados 52 tumores primarios y ocho metástasis: infiltraciones medulares (n = 2), ganglionares (n = 2), ósea (n = 1), intestinal (n = 1), hepáticas (n = 1) y pancreática (n = 1) sin terapia previa. Todos los pacientes recibieron la terapia según los protocolos de SIOP-Europa para alto riesgo.
Histopatología
El diagnóstico histológico, basado en las recomendaciones de la INPC fue revisado por el patólogo del Centro de Referencia Nacional. Fueron analizados el porcentaje de estroma schwanniana, el MKI (bajo, medio y alto siendo < 100, 100 a 200 y > 200 mitosis o cariorrexis por 5.000 células tumorales, respectivamente), el grado de diferenciación y la presencia o ausencia de calcificación.
Análisis genéticos
El análisis genético de los tumores se realizó mediante la técnica de citometría estática (50 casos), de hibridación in situ fluorescente (FISH) (60 casos) y de CGH (24 casos). Para ello se realizaron secciones de las piezas incluidas en parafina, improntas celulares y se extrajo ADN de la misma pieza tumoral utilizada para el estudio histopatológico. Para interpretar los resultados de todas estas técnicas genéticas fue imprescindible conocer el porcentaje de células neuroblásticas en las secciones tumorales. El índice de ADN fue determinado por citometría estática y los resultados se basaron en el Consenso Europeo de Citometría 30. Las improntas celulares con más de 200 células neuroblásticas fueron utilizadas para detectar MNA y para conocer la del de la región 1p36 por FISH. Las sondas utilizadas y las condiciones de hibridación, lavados, detección han sido previamente descritas 31,32. Se utilizó la técnica de CGH 33 con algunas modificaciones. Se analizaron entre 10 y 20 metafases por caso con un microscopio epifluorescente Zeiss axioplan2 con vídeo cámara CCD S31. El análisis de las imágenes se hizo usando el software ISIS v.3.1. Se consideraron los ratios verde/rojo > 1,2 y > 1,5 como número de copias de ADN con ganancia y amplificación, respectivamente, y ratios < 0,8 como pérdidas del número de copias de ADN.
Resultados
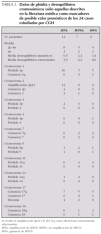
El análisis por FISH del oncogén MYCN y de la integridad de la región 1p36 en los 60 casos estudiados permite dividir la serie tumoral en tres grupos. Grupo 1: MNA, con del 1p36 (n = 29) y no del 1p36 (n = 2); grupo 2: no amplificación de MYCN (MNNA) con del 1p36 (n = 7) y no del 1p36 (n = 14); grupo 3: ganancia del gen MYCN (MNG) con del 1p36 (n = 6), no del 1p36 (n = 1) y desequilibrio de 1p36 (n = 1). Los datos clínicos, histopatológicos y de ploidía relacionados con dichos grupos están resumidos en la tabla 1. Todas las anomalías genéticas diagnosticadas por FISH se confirman en los 24 casos analizados con la técnica de CGH. El 58 % de los casos (14/24) presentan asociados MNA o MNG, del 1p36 y +17q o presencia de cromosoma 17 en disomía (fig. 1). En la tabla 2 se presenta la asociación de los grupos 1, 2 y 3 con los resultados de ploidía, número de desequilibrios cromosómicos y alteraciones cromosómicas presentes.
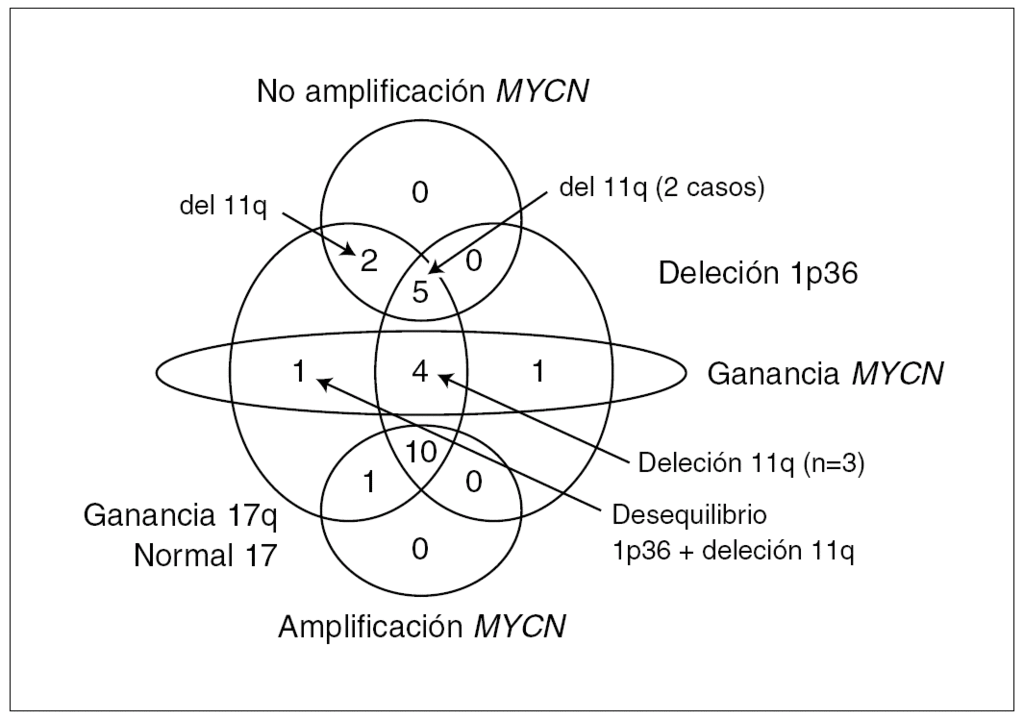
Figura 1. Relaciones entre MYCN, 1p36, cromosoma 17 y 11q en los 24 casos analizados por CGH.
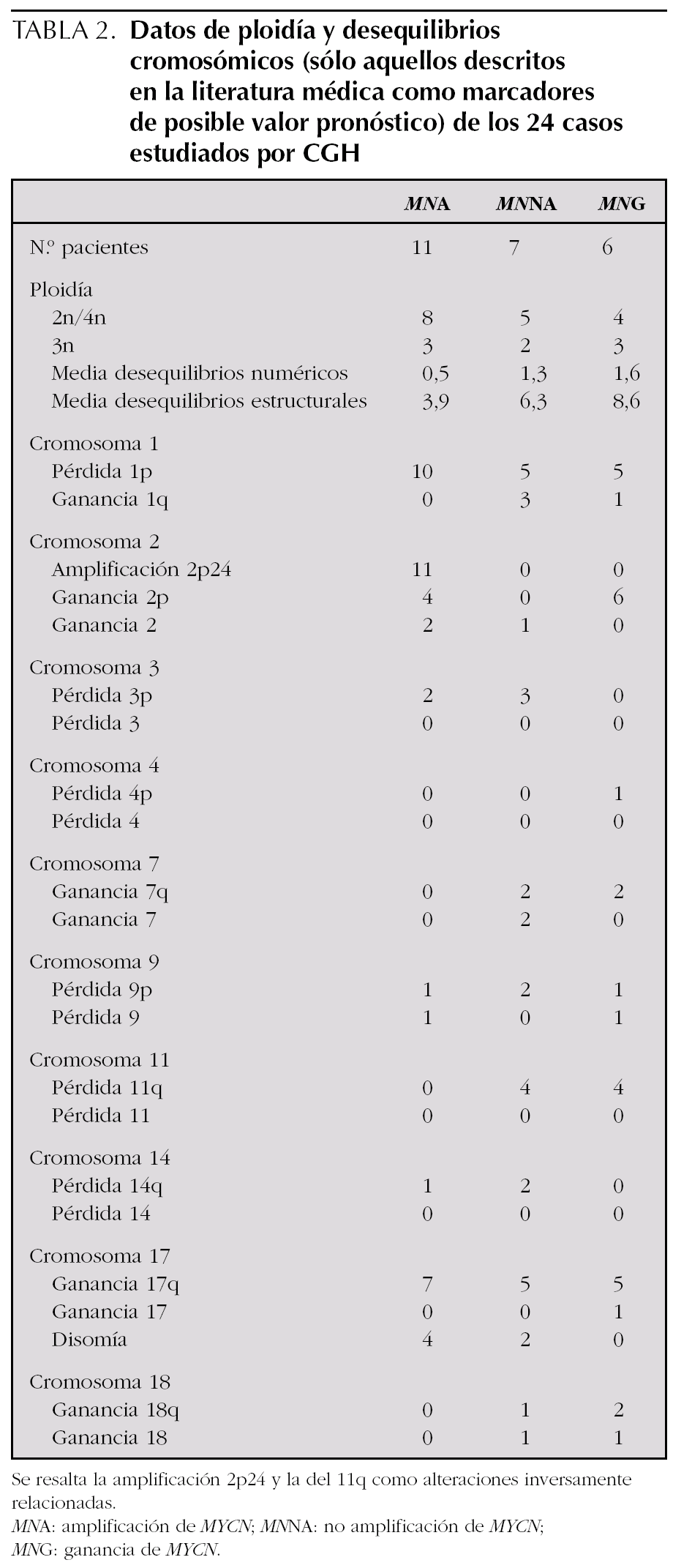
La MNA (fig. 2) presente en 11 casos (45,8 %) se asocia a del 1p36 en aproximadamente el 91 % (10/11) y +17q (63 %; 7/11) o cromosoma 17 en disomía (36 %, 4/11). La MNA (región 2p24) está asociada a ganancia de otras regiones del brazo corto del cromosoma 2 (p21-23) o ganancia completa del cromosoma 2. La del en 1p se limita a la región 1p36 en un caso y es mayor en el resto de casos (en tres de ellos es completa). Únicamente en un caso el cromosoma 1 está sin del. Estos casos MNA presentan pocas ganancias y pérdidas completas cromosómicas y un número escaso de reestructuraciones parciales del resto de cromosomas (media de 3,9 reestructuraciones por tumor). Un caso presenta del 3p22 y en otro caso se observa una del 14q31 estando ausente en todos los casos la del 11q. Ocho de estos casos son diploides y tres triploides. Todos los casos según la INPC presentan un pronóstico histológico desfavorable y todos excepto uno presentan un alto MKI junto con una alta expresión de Ki67. La edad media al diagnóstico es de 39,09 meses. A pesar de que todos los enfermos excepto uno respondieron inicialmente al tratamiento, fallecieron 5 enfermos por progresión y uno por complicaciones.
Figura 2.Ideograma de amplificaciones (barras gruesas a la derecha de cada idiograma cromosómico), ganancias (barras finas a la derecha de cada idiograma cromosómico), y pérdidas (barras a la izquierda de cada idiograma cromosómico) en el grupo de los casos con amplificación del gen MYCN.
La MNNA (fig. 3) presente en 7 casos (29 %) se asocia a del 1p36 y a +17q en el 71 % de los casos (5/7). La del 1p se limita en todos los casos a la región 1p36 y se asocia en 3 casos a ganancias parciales de regiones en 1q, un caso presenta una del intersticial 1p33-35 y otro caso presenta el brazo 1p íntegro. Las ganancias y pérdidas cromosómicas parciales son frecuentes en estos casos MNNA (media de 6,3 reestructuraciones por tumor), entre ellas la del 11q14-23 está presente en 4 casos, en dos de ellos asociada a del 3p21 y en los otros 2 casos asociada a del 14q31. Un caso presenta del 3p21 sin del 11q. Cinco de estos casos son diploides/tetraploides y dos triploides (ambos casos presentan del 11q). El pronóstico histológico en los 4 casos valorables es desfavorable aunque únicamente 2 casos presentan un alto MKI junto con una alta expresión de Ki67. La edad media al diagnóstico es de 51,57 meses. Todos los enfermos excepto uno respondieron inicialmente al tratamiento y 2 enfermos fallecieron por progresión. Dos casos se encuentran en progresión de su enfermedad.

Figura 3.Ideograma de ganancias (barras a la derecha de cada idiograma cromosómico), y pérdidas (barras a la izquierda de cada idiograma cromosómico) en el grupo de los casos sin amplificación del gen MYCN.
Por último, la MNG (fig. 4) presente en 6 casos (25 %), se asocia a del 1p36 y a +17q en 5 casos (83 %); un caso no presenta del 1p36 sino desequilibrio de 1p36, y en otro caso se aprecia una ganancia completa del cromosoma 17. Cuatro casos presentan deleciones en 11q sin estar asociada a del 3p ni del 14q. Las ganancias y pérdidas cromosómicas parciales son frecuentes en todos los casos con MNG (media de 8,6 reestructuraciones por tumor). Cuatro de estos casos son diploides/tetraploides y dos triploides (uno de estos casos presenta del 11q). El pronóstico histológico en los cuatro casos valorables es desfavorable con un alto MKI y una alta expresión de Ki67. La edad media al diagnóstico se encuentra en 44.17 meses. Tres casos no responden al tratamiento, encontrándose en progresión de su enfermedad junto con un cuarto enfermo que tras responder inicialmente al tratamiento se encuentra en la misma situación.
Figura 4. Ideograma de ganancias (barras a la derecha de cada idiograma cromosómico), y pérdidas (barras a la izquierda de cada idiograma cromosómico) en el grupo de los casos con ganancia del gen MYCN
Discusión
La clasificación de riesgo dentro del neuroblastoma se basa actualmente en la determinación de escasos factores clínicos y biológicos 1,23. A pesar de permitir una marcada mejora terapéutica, los datos sugieren un modelo hipotético donde el riesgo podría depender también del momento en que la célula tumoral adquiere cambios genéticos específicos. El objetivo principal de este estudio es la valoración más precisa del riesgo en el momento del diagnóstico en el grupo de NB-HR, sobre la base de nuevos cambios genéticos específicos y por tanto con la detección de mayor número de anomalías genéticas.
Utilizando FISH y de citometría estática se observa, en consonancia con estudios previos, que los 60 NB-HR recibidos en un período de 3 años en el Centro de Referencia Nacional, son tumores en progresión con características genéticas heterogéneas. Por este motivo, se plantea aplicar la CGH metafásica, a un pequeño grupo de estos tumores para valorar la posibilidad de detectar nuevas diferencias genéticas, definir subtipos tumorales basados en estratificación genética y por tanto aplicar al diagnóstico esta técnica en el Centro de Referencia. Con el uso del análisis de CGH metafásico se detectan nuevas alteraciones genéticas, a pesar de que esta técnica tiene un límite de resolución de 10-20 Mb 34,35. Si se clasifica los tumores únicamente según el estatus de MYCN, se observa que +17q es la anomalía más frecuente compartida por los tres subgrupos seguida por la del 1p36. Recientemente Vandesompele et al 19 han detectado que en los neuroblastomas favorables, la presencia del cromosoma 17 en disomía representa un signo de alto riesgo equiparable a +17q; por esta razón se engloba este dato en nuestro estudio junto con +17q. Así mismo, en los grupos de MNNA y MNG se observa tumores que presentan del 11q, hallazgo ausente en el grupo de tumores MNA. Debido a que los estudios realizados mediante micromatrices de CGH con BAC, ADNc u oligonucleótidos, que pueden detectar alteraciones genéticas y de expresión con una mayor resolución, están permitiendo la identificación en neuroblastoma de patrones de expresión génica correlacionadas con desequilibrios genéticos específicos, en particular con del 11q 36,37, se considera esta alteración como identificativa de un subgrupo tumoral. Así, en este estudio se puede diferenciar dos grupos genéticos claros, tumores MNA y tumores con del 11q.
El grupo 1 se identifica y define por la presencia de MNA sin detectar otros hallazgos genéticos específicos. Se observa la integridad de 11q. Presentan muy pocas alteraciones genómicas, indicando que MNA es suficiente para determinar en estos tumores un fenotipo agresivo, aunque ocurren otros cambios genéticos, incluidos del 1p36 y +17q. La del 1p asociada a MNA afecta a regiones amplias y tiene como consecuencia la pérdida de múltiples genes. Dentro de los NB-HR los pacientes de este grupo presenta una media de edad al diagnóstico menor (39,09 meses) y progresan rápidamente. El grupo 2 presenta mayor número de anormalidades estructurales, y está presente entre éstas la del 11q. Estos casos están incluidos tanto en el grupo MNNA como en el grupo de MNG. Por tanto se considera que los tumores con del 11q forman un subgrupo diferenciado de tumores que a su vez pueden o no presentar asociada MNG, del 1p, del 3p o del 14q. Los tumores incluidos en este subgrupo crecen más despacio, la media de edad al diagnóstico es mayor (52 meses) y por tanto acumulan mayor número de anormalidades genéticas secundarias, presumiblemente por el tiempo más largo de evolución. El grupo de tumores que no presentan MNA ni del 11q sigue siendo un grupo genéticamente heterogéneo, en donde no se puede detectar con la técnica de CGH metafásica anomalías comunes identificativas, aunque sí están presentes la +17q y/o el cromosoma 17 en disomía.
Como conclusión, se identifica en este estudio utilizando la CGH metafásica, dos grupos genéticos diferenciados que pueden tener implicaciones diagnósticas y terapéuticas importantes. A pesar de tratarse de un reducido número de casos analizados y con un corto período de seguimiento, ha sido posible definir cambios genéticos asociados a los distintos subgrupos de pacientes. Estos resultados ponen de manifiesto la necesidad de ampliar el estudio a un mayor número de pacientes para definir y establecer los factores de riesgo biológico mediante esta técnica y así garantizar su utilidad en la aplicación clínica junto con las pruebas diagnósticas actuales.
Agradecimientos
Los autores agradecen la colaboración de los técnicos de E. Alonso y J. Benavent, las doctoras D. Sánchez-Izquierdo y J. Cruz así como al estudiante predoctoral M. Enciso en este trabajo.
Financiado por el fondo de investigación sanitaria, proyectos: FIS G03/089 y PI04/0822, Madrid.
Correspondencia: Dra. Rosa Noguera Salvá.
Laboratorio de Patología Molecular. Departamento de Patología
de la Facultad de Medicina y Odontología. Universidad de Valencia.
Avda. Blasco Ibáñez, 17. 46010 Valencia. España.
Correo electrónico: rosa.noguera@uv.es
Recibido en octubre de 2005.
Aceptado para su publicación en febrero de 2006
