


La osteopetrosis maligna infantil (MIM: 259700) es una rara displasia ósea caracterizada por un aumento progresivo de la densidad ósea por alteración en la función de los osteoclastos. Se transmite por herencia autosómica recesiva, con una incidencia de 1:200.000 recién nacidos, con presentación precoz intraútero o en los primeros meses de vida.
Los rasgos típicos de la enfermedad son retraso del crecimiento lineal, problemas dentales, fracturas patológicas, osteomielitis e hipocalcemia debidas a la disminución de la reabsorción ósea; alteraciones hematológicas (anemia, plaquetopenia, leucoeritrobastosis) por invasión de espacios medulares, con hepatoesplenomegalia compensadora; aumento en la frecuencia de infecciones por alteración de la función de monocitos y neutrófilos, y disfunción de pares craneales (ceguera, sordera, parálisis facial) por compresión de los orificios de los nervios craneales. Las radiografías esqueléticas revelan esclerosis ósea difusa.
La osteopetrosis es una entidad heterogénea con varios genes identificados; el 50% de los casos se debe a mutaciones en el gen TCIRG1, siendo menos frecuentes otras mutaciones en otros genes (CLCN7, OSTM1, RANK, RANKL)1,2.
Las características clínicas y radiológicas establecen el diagnóstico y el estudio genético aporta información adicional sobre el pronóstico, la respuesta al tratamiento y la recurrencia de la enfermedad. Sin tratamiento, la evolución natural es progresiva y fatal3.
Describimos el caso de una lactante de 5 meses de edad remitida para estudio por exoftalmos bilateral y fontanela a tensión. El embarazo transcurrió sin incidencias y el parto fue a término, con un Apgar 9/10 y un peso de 3.100 g. Padre y madre desconocen si son consanguíneos y viven sanos. Presenta aspecto desnutrido (talla y peso < p3), con macrocefalia y deformidad craneal por prominencia frontal y occipital. Destacan suturas marcadas con fontanela abombada, proptosis bilateral, nistagmo horizontal y midriasis media sin seguimiento ocular a la luz. El abdomen está distendido, con hepatoesplenomegalia de consistencia dura hasta ambas fosas iliacas. Soplo sistólico I/VI. Retraso psicomotor moderado, con tono y fuerza normal.
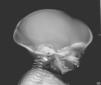
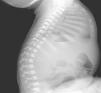
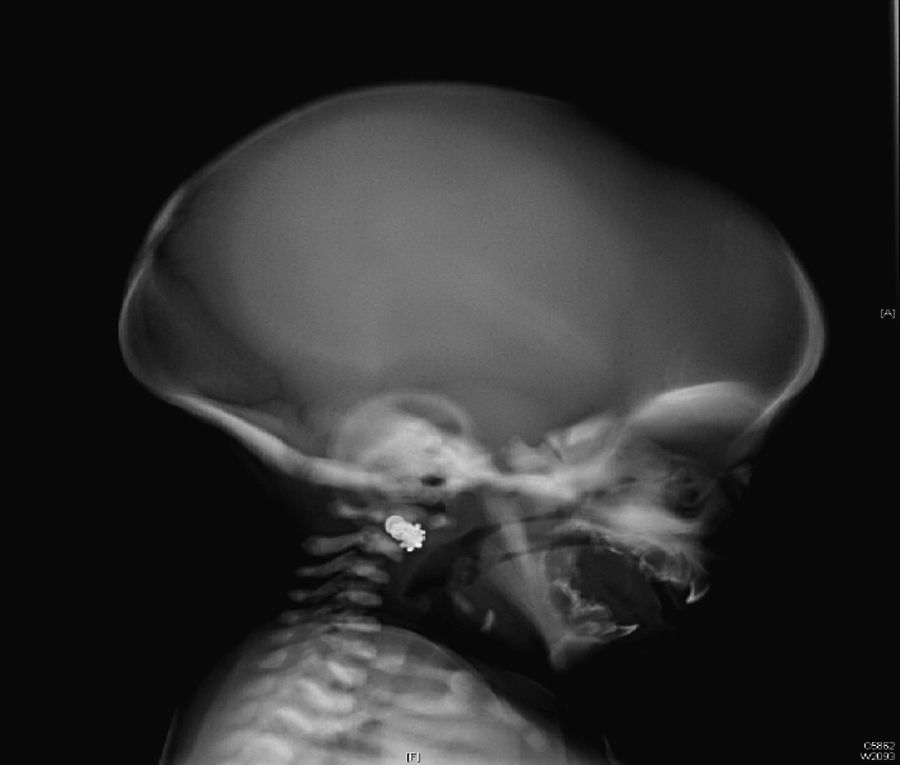
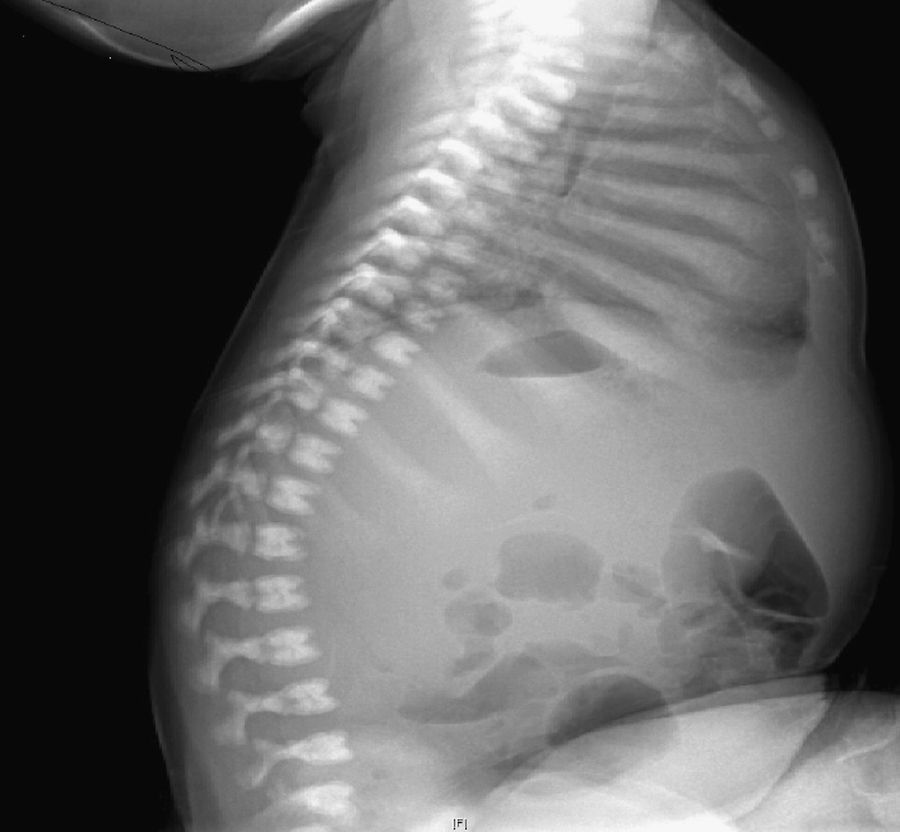
Inicialmente, se realiza una radiografía craneal donde se observan esclerosis y engrosamiento de la base del cráneo (fig. 1). Ante estos hallazgos, ampliamos el estudio radiológico, evidenciándose un aumento difuso de la densidad ósea (fenómeno hueso dentro de hueso), llamativo a nivel de los cuerpos vertebrales (fig. 2), esclerosis de costillas y huesos de la pelvis, y metáfisis en «maza» de huesos largos, donde no se reconoce cavidad medular.
Ante estas imágenes radiológicas compatibles con displasia ósea, se procede a realizar las siguientes pruebas: análisis sanguíneo (Hb: 8,0 g/dl; hematocrito: 24,8%; leucocitos 31.600/μl [neutrófilos 33%, linfocitos 47%, monocitos 10%, eosinófilos 2%, basófilos 0%]; eritroblastos 5%; células inmaduras 1%; plaquetas 73.000/μl; calcio 8,37 mg/dl; fósforo 2,92 mg/dl; fosfatasa alcalina: 950 U/l; PTH 220 pg/ml), ecografía abdominal (hepatoesplenomegalia); ecografía cerebral (alteración de la ecogenicidad de sustancia blanca subcortical); potenciales evocados visuales (ausentes); potenciales auditivos (hipoacusia bilateral de 50 dB); RM cerebral (significativo engrosamiento de la calota en base del cráneo), y TC craneal (órbitas pequeñas por aumento de densidad ósea, proptosis ocular bilateral, agujeros ópticos permeables, cajas timpánicas bien configuradas).
En la anamnesis detallada descubrimos la presencia de un sobrino de la madre afectado de osteopetrosis maligna infantil (mutación del gen TCIRG1) y en el árbol genealógico existe la posibilidad de herencia autosómica recesiva al estar ambas familias unidas hace tres generaciones.
Ante el diagnóstico clínico y radiológico de osteopetrosis, se realiza un estudio genético que confirma la mutación del gen TCIRG1 y solicitamos las pruebas necesarias para realizar el transplante de progenitores hematopoyéticos. Como tratamiento inicial, se administran corticoides orales (1 mg/kg/día) e interferón gamma 1b (1,5 μg/kg/dosis, 3 veces/semana)4, realizándose controles analíticos y clínicos de la paciente. Tras una semana de tratamiento con corticoides, presenta buena respuesta hematológica (Hb de 9,8 y 187.000 plaquetas), que permite mejorar su estado clínico. Actualmente, recibe tratamiento con interferón gamma 1b, esperando el transplante. No ha presentado infecciones ni hipocalcemia y ha precisado transfusiones de concentrados de hematíes mensuales. El estudio de histocompatibilidad con los progenitores no es viable, por lo que se realizará un transplante de cordón umbilical con un HLA compatible no emparentado.
El trasplante de progenitores hematopoyéticos es el único tratamiento que puede ser definitivo, con un porcentaje de supervivencia del 74 al 43%, dependiendo del grado de histocompatibilidad del donante5,6. Destacamos que los casos que más se benefician del trasplante son los causados por mutaciones en el gen TCIRG1 y, en menor medida, aquellas mutaciones del gen CLCN7, siendo el beneficio dudoso para el resto de los genes7. El tratamiento con interferón gamma 1b, corticoides y calcitriol puede ser útil en aquellos pacientes que no se benefician del trasplante o hasta que este sea posible.
