



La enfermedad inflamatoria intestinal (EII) puede acompañarse de manifestaciones extraintestinales (MEI) prácticamente en la totalidad de los órganos y sistemas. Se estima una prevalencia variable entre el 6 y el 47% en adultos con EII. De entre éstas, las más frecuentes son las musculoesqueléticas, seguidas de las dermatológicas. Pese a que en las poblaciones pediátricas los datos disponibles son limitados, algunos estudios indican que la prevalencia podría ser incluso mayor1. A continuación se presentan varios casos de pacientes pediátricos afectados de EII que han presentado diferentes manifestaciones cutáneas asociadas.
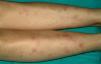
Caso 1. Paciente mujer de 13 años con diarrea, abdominalgia y episodios febriles de 3 semanas de evolución. Analíticamente presenta marcadores inflamatorios elevados. La colonoscopia muestra hallazgos indicativos de enfermedad de Crohn (EC) ileocolónica y la anatomía patológica confirma el diagnóstico, por lo que se administra tratamiento con nutrición enteral exclusiva con fórmula polimérica y mesalazina oral. A los 3 días de iniciar el tratamiento aparecen lesiones eritematosas, induradas y dolorosas en la zona pretibial de ambas extremidades inferiores (fig. 1). Las biopsias de estas lesiones muestran paniculitis lobular, lo que confirma la sospecha clínica de eritema nudoso, y se inicia un tratamiento tópico con yoduro potásico. A las 2 semanas del inicio de la nutrición enteral, ante la ausencia de respuesta clínica (PCDAI [paediatric Crohn's disease activity index‘índice de actividad de la enfermedad de Crohn pediátrica’]: 27,5), la persistencia de fiebre y signos analíticos de inflamación así como la coexistencia de eritema nudoso, se reevalúa la situación y se decide iniciar tratamiento con infliximab (5 mg/kg) en dosis en pauta de ataque (0, 2 y 6 semanas) y azatioprina. La paciente presenta respuesta tras la primera dosis, con resolución total de la lesión cutánea y normalización clínica y ganancia ponderal tras la segunda dosis (PCDAI: 12,5), y entra en remisión tras la tercera dosis (PCDAI: 0). La paciente sigue tratamiento con azatioprina (2,5 mg/kg/día), se mantiene en remisión clínica y sin reaparición de las lesiones cutáneas. A los 3 años y medio de la última dosis de infliximab presenta reaparición de su sintomatología digestiva. Se reinicia infliximab en nueva tanda de ataque y posterior mantenimiento cada 8 semanas lográndose la remisión completa. Un año más tarde, en tratamiento combinado (infliximab y azatioprina) y en remisión completa, es dada de alta de la Sección por mayoría de edad y es transferida a una Unidad de EII del adulto.
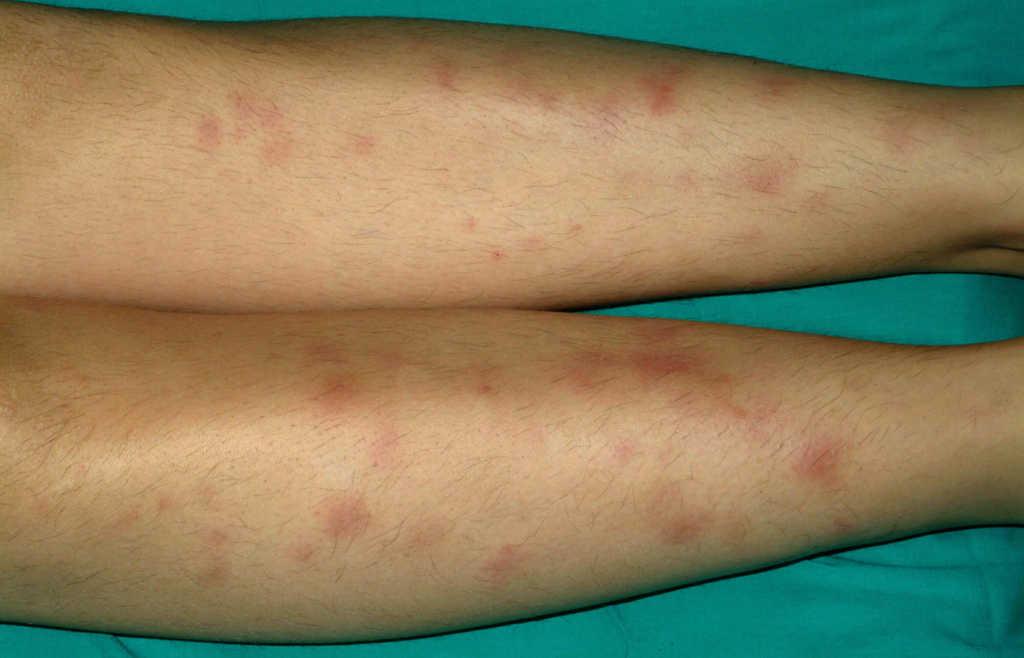
Figura 1. Eritema nudoso en la zona pretibial de ambas extremidades inferiores y de aparición al comienzo de la enfermedad de Crohn.
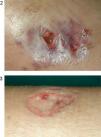
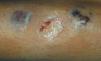
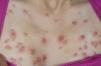
Caso 2. Paciente mujer de 15 años con diarreas, ocasionalmente sanguinolentas, y dolor abdominal de 2 meses de evolución, pérdida de peso y episodios febriles. Refiere amenorrea secundaria desde hace 2 meses y aparición de aftas orales en los últimos días. Presenta anemia ferropénica y marcadores inflamatorios elevados. La colonoscopia muestra lesiones indicativas de Crohn colónico, con íleon terminal preservado. Se administra tratamiento con nutrición enteral y azatioprina. A las 24 h de iniciar el tratamiento aparece una lesión única eritematosa y dolorosa, de 1 cm de diámetro en la región pretibial izquierda. Ante la sospecha inicial de eritema nudoso incipiente, se decide mantener una conducta expectante. Se produce rápida evolución de la lesión, que en 48 h aumenta de tamaño, toma aspecto pustuloso, con supuración serohemática en la zona central ulcerada y zona periférica tumefacta de aspecto violáceo (figs. 2 y 3). La biopsia cutánea muestra dermatosis neutrofílica compatible con pioderma gangrenoso, por lo que se añade tratamiento tópico con esteroides. En los días posteriores aparecen 2 nuevas lesiones satélites eritematosas próximas (fig. 4) que progresivamente evolucionan a formas ulceradas. Se observa fenómeno de patergia en la zona de punción y biopsia previa. Paralelamente, se evidencia empeoramiento progresivo de su sintomatología digestiva así como aparición de nuevas lesiones en la mucosa oral. Ante el fracaso del tratamiento administrado y la presencia de las lesiones cutáneas, se decide iniciar tratamiento con infliximab, sin que se observe respuesta significativa. Persiste el mal estado general, con fiebre alta y leucocitosis importante (20,8 × 109/l, el 89% de neutrófilos). Aparecen nuevas lesiones múltiples diseminadas, de predominio en el tórax y el cuello, de aspecto papular, con halo eritematoso, de alrededor de 1 cm de diámetro (fig. 5), por lo que a la semana se inicia tratamiento con prednisona por vía intravenosa (1 mg/kg/día). Se descartan procesos infecciosos intercurrentes, tanto sistémicos como cutáneos (frotis y cultivos estériles). La histología de las nuevas lesiones muestra denso infiltrado neutrofílico perivascular, sin signos de vasculitis, hallazgos compatibles con síndrome de Sweet. Se produce progresiva mejoría parcial y estabilización de las lesiones. En este contexto presenta clínica brusca de disnea, taquipnea, estridor inspiratorio y tiraje supraclavicular, pero mantiene buena oxigenación. Se realiza fibrobroncoscopia en la que se observan múltiples lesiones pustulosas sobre la mucosa friable en tráquea y bronquios principales. Dadas las características de la mucosa, se decide no realizar biopsias. El lavado broncoalveolar muestra inflamación aguda con predominio de leucocitos polimorfonucleares. Ante la sospecha de afectación neutrofílica del árbol respiratorio en el contexto de síndrome de Sweet se cambia el tratamiento a bolos de metilprednisolona (1 g). Se realiza tomografía computarizada torácica, en la que descarta lesiones pulmonares compatibles con alveolitis neutrofílica, y espirometría, que es normal. La paciente experimenta clara mejoría tras la administración del primer bolo y la dificultad respiratoria desaparece. Se administran otros 2 bolos y se pasa posteriormente a metilprednisolona (20 mg/12 h). Presenta buena evolución posterior, con ausencia de sintomatología digestiva, y estabilización y reepitelización lenta de las lesiones cutáneas, que habían cubierto progresivamente gran parte de la superficie corporal. Es dada de alta con pauta de descenso de esteroides, y con azatioprina y calcio oral. En una nueva broncoscopia al mes del alta se observa franca mejoría de las lesiones. La paciente presenta buena evolución, por lo que se suspende la corticoterapia 2 meses tras el alta. Actualmente se encuentra en remisión y en tratamiento con azatioprina. Las lesiones cutáneas han dejado múltiples y evidentes cicatrices queloides diseminadas (fig. 6).

Figuras 2 y 3. Lesión inicial de pioderma gangrenoso rápidamente evolutiva.
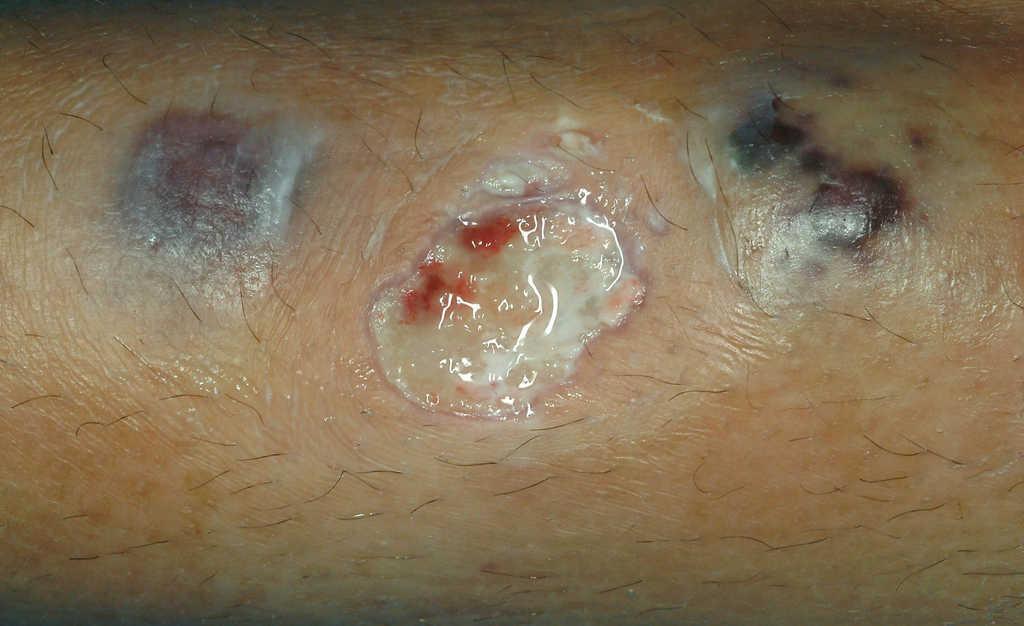
Figura 4. Aparición de lesiones satélites próximas a la lesión inicial en la paciente número 2.
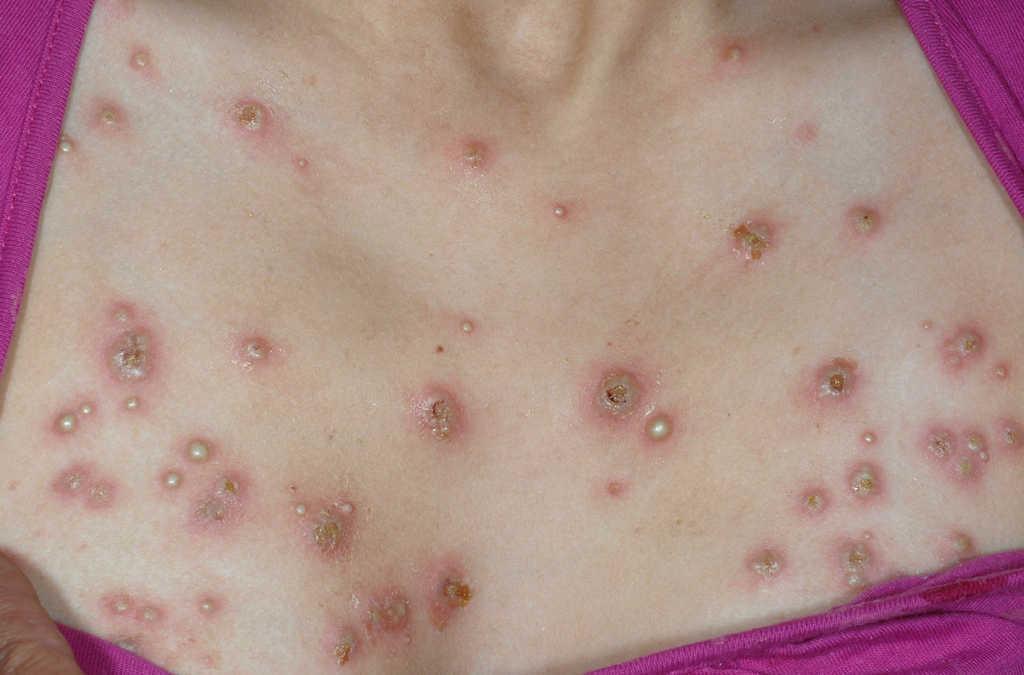
Figura 5. Lesiones múltiples de predominio en el tórax indicativas de síndrome de Sweet.
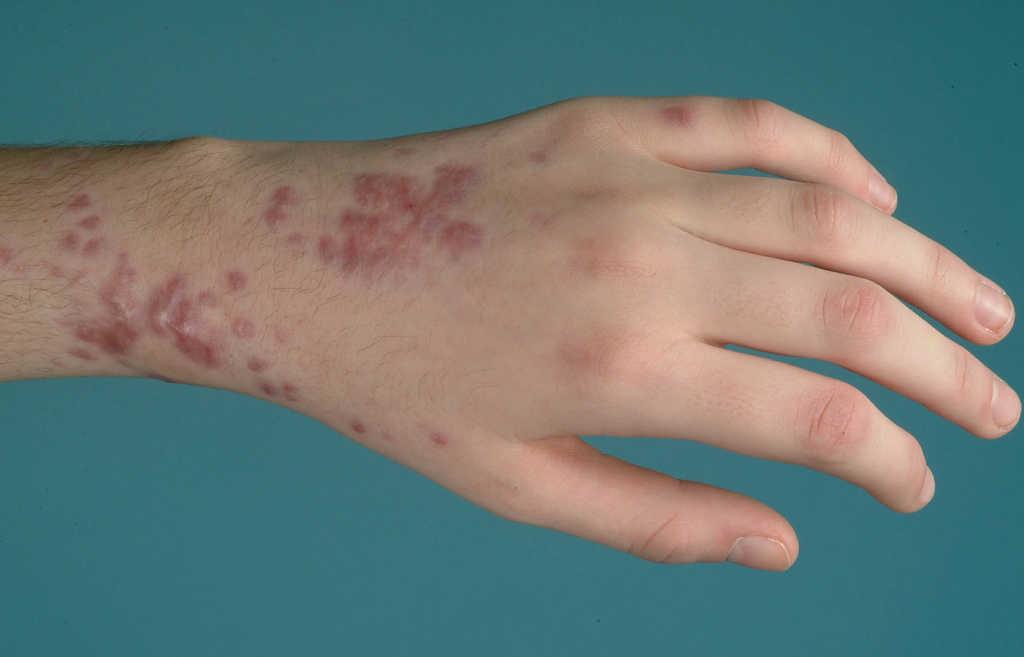
Figura 6. Lesiones residuales diseminadas persistentes en la paciente número 2.
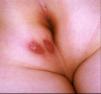
Caso 3. Paciente varón de 10 años con diarrea, abdominalgia y pérdida de peso de 2 meses de evolución. La colonoscopia muestra EC ileocólica, y se inicia tratamiento con nutrición enteral exclusiva y mesalazina. Presenta buena respuesta y se logra la remisión. Al año del diagnóstico reaparece la sintomatología, por lo que se inicia tratamiento con budesonida, con buena respuesta. Se mantiene la remisión posterior con mesalazina. Tras un año y medio en esta situación, consulta por la aparición de 2 lesiones eritematosas en el glúteo izquierdo (fig. 7), que coincide con un nuevo episodio de diarreas. Se realiza resonancia magnética que descarta lesiones abscesificadas o trayecto fistuloso subyacente. Las biopsias cutáneas revelan la presencia de granulomas, y se establece el diagnóstico de Crohn metastásico. Se inicia tratamiento con esteroides tópicos y mediante infiltración, sin que se logre la resolución completa de la lesión cutánea, que presenta fases supurativas. Se inicia la administración de corticoides sistémicos y azatioprina, se logra la mejoría y se puede llegar posteriormente a la suspensión total de esteroides. Durante los 3 años siguientes al inicio de la administración de azatioprina, dada la evolución tórpida de su cuadro digestivo, el paciente requiere varias tandas de budesonida. Ante esta situación se decide tratamiento con infliximab; el paciente presenta clara mejoría y se obtiene la remisión mantenida en régimen de tratamiento combinado con azatioprina e infliximab cada 8 semanas durante un año, sin que reaparezcan de las lesiones glúteas.
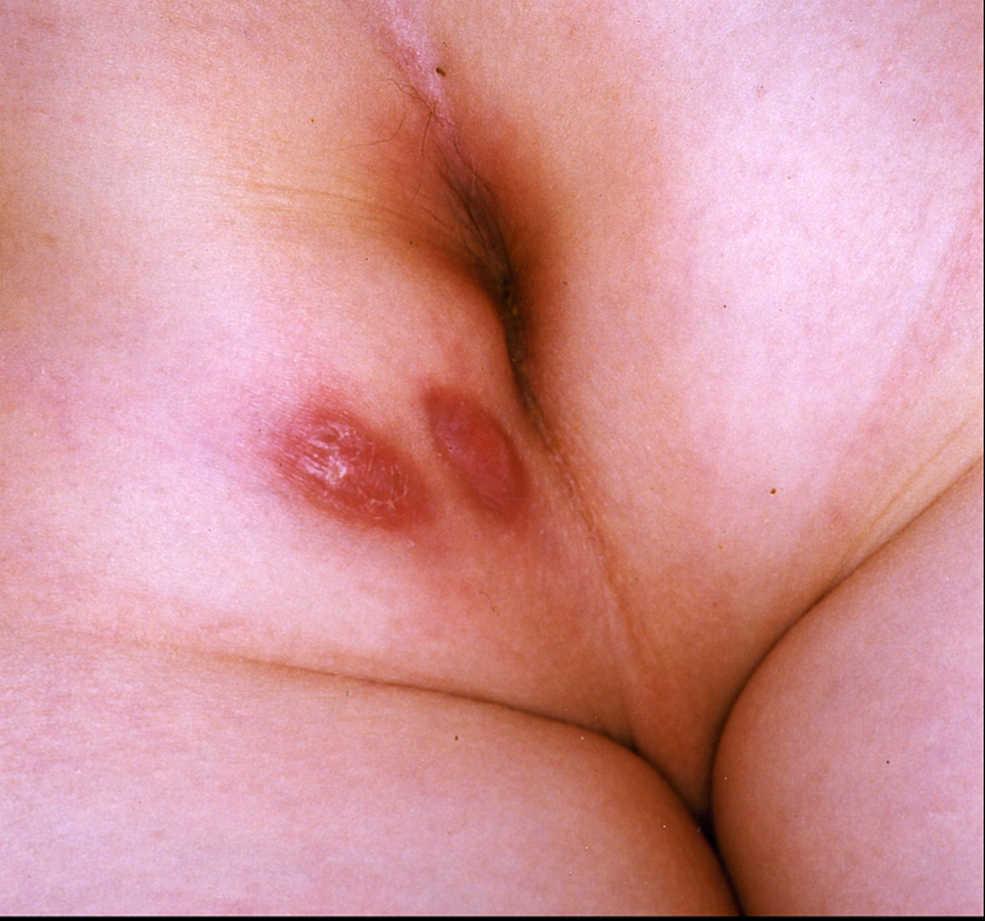
Figura 7. Enfermedad de Crohn metastásica en el glúteo izquierdo en el paciente número 3.
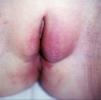
Caso 4. Paciente mujer de 10 años que consulta a Dermatología por aparición de edema y eritema en labios mayores de 4 meses de evolución (fig. 8). Antecedente de fisura anal acompañada de hemorragia ocasional desde los 5 años de edad, sin alteración del ritmo intestinal. Las biopsias muestran dermatitis granulomatosa por lo que se la envía a Gastroenterología para la valoración de posible EC. La familia cuenta episodios febriles autolimitados durante el año previo y pérdida de peso no cuantificado en los últimos meses. A la exploración se observa lesión perianal supurativa indicativa de fístula enterocutánea. La analítica muestra marcadores inflamatorios elevados, discreta anemia y trombocitosis. La colonoscopia muestra lesiones aftosas y úlceras en el ciego, la válvula ileocecal y el íleon terminal indicativas de EC ileocecal. Se inicia tratamiento con mesalazina, metronidazol y corticoides tópicos en la vulva, con lo que se recupera el estado nutricional, se cierra prácticamente por completo la fístula y las lesiones vulvares mejoran parcialmente. Al mantenimiento posterior con mesalazina se añaden periódicamente tandas de corticoides tópicos por evolución tórpida de la lesión cutánea. A partir de los 2 años tras el diagnóstico, presenta diversos brotes con sintomatología digestiva, reactivación de su enfermedad perianal y empeoramiento de sus lesiones vulvares, por lo que precisa tratamiento con esteroides sistémicos. Por situación de dependencia a los corticodes, se administra tratamiento con azatioprina, y se consigue temporalmente la remisión libre de esteroides. Tras 3 años de remisión con azatioprina y mesalazina, presenta reactivación de su enfermedad digestiva, perianal y vulvar, por lo que se inicia tratamiento con infliximab, con buena respuesta inicial. Se establece pauta de mantenimiento posterior con dosis cada 8 semanas y azatioprina, con lo que la remisión se mantiene.
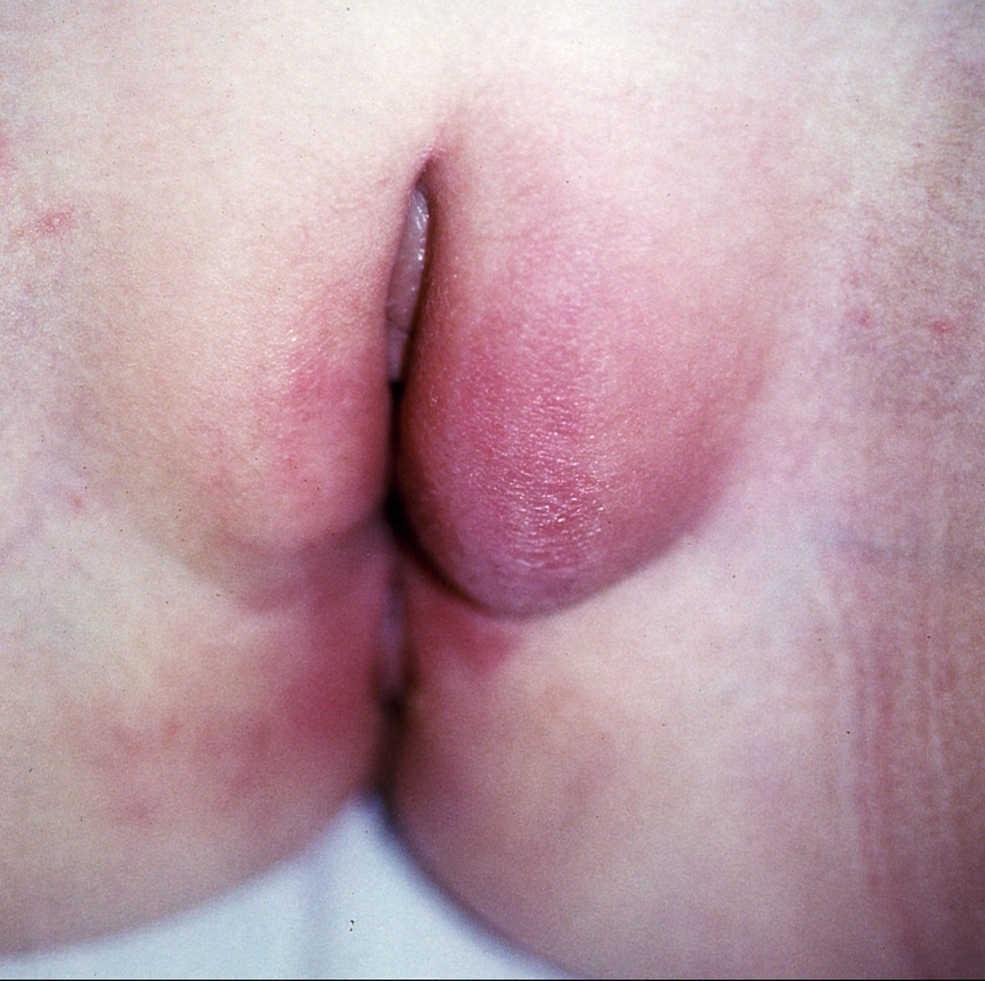
Figura 8. Enfermedad de Crohn metastásica vulvar en forma de edema y eritema de los genitales externos en la paciente número 4.
DiscusiónLa EII (EC, colitis ulcerosa [CU], colitis indeterminada) puede acompañarse de MEI en diferentes órganos y sistemas (tabla 1). Los datos comunicados varían según las series, si bien estudios recientes estiman una prevalencia de al menos una MEI en pacientes adultos entre el 6 y el 47%2,3. Las más frecuentes son las musculoesqueléticas (20%), seguidas de las dermatológicas (del 10 al 15%).
Tabla 1. Manifestaciones extraintestinales de la enfermedad inflamatoria intestinal
| MEI relacionadas con la inflamación | MEI secundarias a extensión directa o a complicación de la EII |
| Dermatológicas | |
| Eritema nudoso | Nefrolitiasis |
| Pioderma gangrenoso | Uropatía obstructiva |
| Pioestomatitis vegetans | Fístula |
| Psoriasis | Pancreatitis |
| Eritema multiforme | Otras: |
| Epidermolisis ampollosa acquista | Hematológicas |
| Síndrome de Sweet | Anemia |
| Enfermedad de Crohn metastásica | Leucocitosis |
| Oftalmológicas | Trombocitosis |
| Uveitis e iritis | Trombocitopenia |
| Episcleritis | Trastornos de la coagulación |
| Escleritis | Hipercoagulabilidad |
| Conjuntivitis | Trombosis de vena porta |
| Glaucoma | Cardiológicas |
| Enfermedad vascular de la retina | Pleuropericarditis |
| Musculoesqueléticas | Miocardiopatía |
| Artropatía periférica | Endocarditis |
| Pauciarticular | Miocarditis |
| Poliarticular | Pulmonares |
| Artropatía axial | Alveolitis fibrosante |
| Espondilitis anquilosante | Vasculitis pulmonar |
| Orales | Fibrosis apical |
| Estomatitis angular | Bronquiectasias |
| Estomatitis aftosa | Bronquitis |
| Piostomatitis vegetans | Bronquiolitis |
| Estenosis traqueal | |
| Hepatobiliares | Enfermedad pulmonar granulomatosa |
| Cirrosis | Anomalías en la función pulmonar |
| Colangiocarcinoma | Neurológicas |
| Esteatosis | Neuropatía |
| Hepatitis granulomatosa | Miopatía |
| Colelitiasis | Meningitis |
| Hepatitis autoinmune | Convulsiones |
| Absceso hepático | Amiloidosis |
| Colangitis esclerosante primaria | Cáncer extraintestinal |
| Pericolangitis | Leucemia mieloide aguda |
MEI: manifestaciones extraintestinales.
Las MEI se clasifican en diferentes categorías. Por un lado, aquéllas relacionadas con la actividad inflamatoria intestinal, como es el caso de las cutáneas, oculares, articulares y orales. Su actividad discurre paralela a la del la enfermedad intestinal y normalmente responden al tratamiento dirigido contra ésta. Por otro lado, hay otro grupo de MEI en que el curso discurre independiente de la enfermedad de base. En este grupo se incluirían las manifestaciones hepáticas (fundamentalmente colangitis esclerosante primaria), vasculares, hematológicas, neurológicas, cardíacas, pulmonares y la amiloidosis. Otro grupo lo constituirían aquellas MEI secundarias a extensión local de la enfermedad primaria, que son más frecuentes en la EC que en la CU: nefrolitiasis, uropatía obstructiva, coledocolitiasis y pancreatitis. Y por último, hay un grupo de MEI de origen iatrogénico, como es el caso de la aplasia medular y la miopatía secundaria a los esteroides. Pese a que algunas revisiones engloban dentro de las MEI a la alteración de la densidad mineral ósea (osteopenia, osteoporosis) y al retraso de crecimiento en la EII pediátrica, ambas serían más bien complicaciones en las que influyen tanto factores propios de la enfermedad como otros factores medicamentosos.
Hay pocos datos de prevalencia de MEI en la EII pediátrica, pero algunos estudios indican que podrían ser incluso más frecuentes que en adultos. Grossman et al detectan la presencia de una MEI en una serie de 41 niños con EII hasta en el 68%4. Por otro lado, Stawarski et al comunican que el 50% de los pacientes pediátricos con CU y hasta el 80% de aquellos pacientes con EC tenían, al menos, una MEI asociada5.
Las manifestaciones cutáneas de la EII pediátrica están presentes al diagnóstico en entre el 1 y el 8% de los pacientes, según datos reportados en Canadá y en Inglaterra6. Clásicamente se han clasificado en específicas, reactivas y secundarias a desórdenes acompañantes o a déficits nutricionales. Aquéllas directamente relacionadas con el proceso inflamatorio de la EC serían las lesiones cutáneas perianales y periostomales, la granulomatosis oral y la EC metastásica. Entre aquellas manifestaciones reactivas, las más frecuentes son el eritema nudoso y el pioderma gangrenoso, si bien pueden aparecer también pioderma vegetante, eritema multiforme, poliarteritis nudosa cutánea, urticaria vasculitis necrosante y síndrome de Sweet. Por último, hay un grupo de manifestaciones mucocutáneas, consecuencia de defectos nutricionales (acrodermatitis enteropática) o de fenómenos autoinmunitarios asociados (psoriasis, lupus eritematoso, esclerodermia, epidermolisis ampollosa, vitíligo, alopecia areata o amiloidosis). Dentro de estas clasificaciones de MEI cutáneas debería omitirse la enfermedad perianal por ser algo más que una MEI cutánea; actualmente está considerada, a partir de la clasificación de Montreal, una entidad diferenciada y con características específicas dentro de la EC7.
El eritema nudoso es la manifestación cutánea más frecuente en EII, es más habitual en la EC que en la CU y aparece habitualmente en momentos de actividad de la enfermedad. La prevalencia estimada en adultos con EII es del 2 al 8%2,8, mientras que en los niños oscila entre el 12,5% en la CU y el 56% en la EC4. Es una paniculitis secundaria a una reacción inmunológica de hipersensibilidad retardada frente a diferentes estímulos antigénicos. El agente causante se llega a identificar en el 40% de los casos (infecciones estreptocócicas, tuberculosis, micosis, fármacos, EII, procesos autoinmunes, neoplasias, etc.), pero habitualmente se presenta de forma idiopática. Su etiología es desconocida, si bien parece asociarse a cierta predisposición genética (región HLA-B15 del cromosoma 6).
El caso 1 ilustra el eritema nudoso clásico asociado a EII; en cuanto a la forma de presentación (nódulos eritematosos, de 1 a 5 cm de diámetro, levemente prominentes, hiperémicos y dolorosos a la palpación, mal delimitados, sin tendencia a la ulceración y de localización preferente en superficies extensoras de extremidades inferiores), al momento de aparición (frecuente al comienzo) y a los hallazgos histológicos (reacción linfocitaria en hipodermis, de predominio septal —paniculitis lobulillar— sin vasculitis). Es más frecuente en mujeres, en afectación colónica extensa y tiende a recurrir en aproximadamente la mitad de los casos. El diagnóstico suele ser clínico y la confirmación anatomopatológica es raramente necesaria. Mientras que el eritema nudoso asociado a otras enfermedades tiende a desaparecer espontáneamente en 3 a 6 semanas con medidas de soporte (reposo, elevación de las extremidades, vendaje compresivo y antiinflamatorio no esteroideo), en los pacientes con EII su curso es más tórpido9. Habitualmente responde al tratamiento de la enfermedad subyacente con esteroides e inmunosupresores. Sobre la base de la experiencia de los autores del presente artículo, previamente comunicada, se decidió iniciar tratamiento con infliximab, con lo que se logra la resolución total de las lesiones, sin que se hayan presentado recidivas posteriores10.
El pioderma gangrenoso es una lesión cutánea crónica de tipo ulcerativo que puede aparecer de forma grave y,en ocasiones, es más incapacitante que la enfermedad subyacente. Se ha descrito en el contexto de diversas enfermedades sistémicas (EII, hepatitis C, artritis reumatoide, mielodisplasia, leucemia). Su prevalencia oscila entre el 0,5 y el 5% de los pacientes con EII11,12, es más frecuente en la CU (entidad en la que aparece con mayor frecuencia) que en la EC. No hay datos de prevalencia específicos en EII pediátrica. Habitualmente afecta a los pacientes con enfermedad activa (90%), con discreta preponderancia del sexo femenino, y en casos de afectación colónica extensa. Algunos casos de presentación previa al diagnóstico de EII, de aparición en fases quiescentes de éstas o incluso tras colectomía también se han comunicado. Se piensa que determinados autoanticuerpos con reactividad cruzada frente a antígenos intestinales desempeñarían un papel importante en su aparición en la EII. Hay 4 formas de presentación: clásica, pustular, ampollosa y vegetativa. La forma clásica aparece como pústula o nódulo que crece y se extiende rápidamente hacia la piel circundante, y se convierte en úlceras penetrantes de centro necrótico y purulento con bordes irregulares y violáceos. Típicamente aparece en la superficie extensora de las extremidades inferiores, como en este caso, pero pueden aparecer también en la cabeza, el cuello, los genitales y la zona periostomal. Hasta en el 70% de los casos aparece en múltiples localizaciones13. La mitad de los pacientes presentan formación de úlceras secundarias a traumatismos en la lesión, fenómeno conocido como patergia. El pioderma gangrenoso pustular se presenta como una pústula aséptica dolorosa que no progresa a úlcera. La forma ampollosa comienza como una ampolla a tensión que rápidamente progresa a úlcera. Por último, la forma vegetante comienza como una úlcera superficial que lentamente se convierte en una lesión exofítica o vegetante. Los subtipos que suelen aparecer en la EII son la forma clásica y la pustular, mientras que las formas ampollosa y vegetante se asocian preferentemente a los procesos mieloproliferativos. La histología muestra vasculitis linfocítica periférica en las zonas eritematosas, infiltración neutrofílica y abscesificación central.
En el caso 2 (pioderma gangrenoso clásico de localización típica y múltiple) hay varios aspectos reseñables: por una lado, como se ha comentado, pese a aparecer más frecuentemente en pacientes con CU, esta paciente presentaba datos endoscópicos e histológicos compatibles con EC, aunque de localización estrictamente colónica. Otro dato para destacar es la coexistencia de pioderma y síndrome de Sweet, combinación de la que hay pocos datos comunicados en la literatura médica14. Algunos autores consideran ambas entidades como diferentes presentaciones de dermatosis neutrofílica15.
Pese a que el pioderma gangrenoso habitualmente se resuelve con el tratamiento de la EII, hay casos de evolución tórpida y recidivante. Algunas formas leves responden a un tratamiento tópico, (esteroides intralesionales, cromoglicato sódico, ácido 5-aminosalicílico). Las formas más graves pueden requerir agentes sistémicos: sulfasalazina, dapsona, esteroides e inmunomoduladores (azatioprina, ciclofosfamida, ciclosporina, metotrexato, micofenolato mofetil y tacrolimus)16. Las técnicas de granulocitoaféresis también han demostrado su eficacia en las formas complicadas. El tratamiento efectivo con infliximab tanto en niños como en adultos se ha comunicado también recientemente 17,18, si bien no fue el caso de esta paciente.
El síndrome de Sweet, o dermatosis neutrofílica febril aguda, se caracteriza por la aparición brusca de una erupción cutánea compuesta por placas o pápulas eritematosas, más o menos dolorosas y bien delimitadas, su localización puede ser variable o generalizada, y se acompaña con frecuencia de fiebre, leucocitosis y artralgia. Las lesiones aparecen preferentemente en la cara, el cuello, el tronco y las extremidades, aunque también se han descrito en los dedos, el conducto auditivo externo y la cavidad oral. Para establecer el diagnóstico de seguridad se requieren una serie de criterios establecidos en 1986 y posteriormente modificados por Driesch en 199419 (tabla 2). Se ha descrito asociado a diferentes procesos, incluidos infecciones, desórdenes hematopoyéticos o linfoides, tumores sólidos, enfermedades reumatológicas, embarazo, toxicidades medicamentosas y EII. Dentro de su excepcionalidad (su asociación a EII es una rareza20), la CU y la EC son los procesos con los que más frecuentemente se relaciona. Generalmente se manifiesta durante exacerbaciones de la enfermedad de base pero puede anticiparse al diagnóstico de ésta. Pese a que se manifiesta básicamente en la piel, puede afectar a otros órganos, como el tracto respiratorio y, en menor medida, a los ojos, los huesos, el páncreas, el hígado, las articulaciones y los riñones. Hay varias series publicadas de pacientes con afectación pulmonar21,22. Los casos descritos de alveolitis neutrofílica presentan sintomatología importante (tos persistente, disnea y derrame pleural) que puede llegar al fracaso respiratorio, con hallazgos radiológicos de condensación parenquimatosa y mala respuesta a antibióticos. La biopsia pulmonar abierta o transbronquial confirma el diagnóstico. Asimismo, hay casos descritos de manifestaciones en el árbol bronquial23, donde se observa la presencia de múltiples lesiones pustulosas en la mucosa, semejantes a las que aparecen en la piel, como fue el caso de esta paciente. La sintomatología respiratoria, asociada a las manifestaciones cutáneas y en el contexto de fiebre de inicio brusco, debe hacer sospechar esta afectación. El lavado bronquioalveolar, que es estéril, confirma el claro predominio de neutrófilos.
Table 2. Criterios diagnósticos propuestos para el síndrome de Sweet
| Criterios mayores |
|
| Criterios menores |
|
| Para un diagnóstico definitivo deben cumplirse los 2 criterios mayores y al menos 2 de los criterios menores. Modificado de: Von den Driesch P. Sweet's syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol 1994;31:535–60. |
Su patogenia se desconoce, pero se postula una cierta predisposición genética asociada a una reacción de hipersensibilidad en la que los neutrófilos desempeñarían un papel importante. Las formas leves suelen curar sin dejar cicatriz tras un período variable de actividad. Se ha descrito recurrencia hasta en el 50% de los casos. Los casos graves suelen responder a esteroides sistémicos. En el caso de Sweet diseminado con afectación de otros órganos y sistemas, como es el caso de la implicación respiratoria, se recomienda el uso de bolos de esteroides. Otros tratamientos utilizados con variable eficacia han sido azatioprina, indometacina, colchicina, dapsona, yoduro potásico y talidomida.
Bajo el término de EC metastásica se denomina la presencia de granulomas no caseificantes en la piel de pacientes con EC, a distancia del tracto intestinal y separado de éste por tejido sano; se excluyen así las lesiones fistulizantes o las complicaciones periostomales. Es una complicación poco frecuente y, mientras en adultos aparece después del diagnóstico inicial en el 70% de los casos, en niños puede aparecer simultáneamente hasta en la mitad de los casos, como fue el caso de la paciente número 4. Se calcula que cuando se observa una EC metastásica aislada, el tiempo de demora hasta que se manifiesta la enfermedad intestinal oscila entre 2 meses y 4 años en adultos, y entre 9 meses y 14 años en niños24. Las formas típicas de presentación también varían según la edad: en adultos suele aparecer en forma de nódulos o placas (únicos o múltiples), ulcerados o no, preferentemente en las extremidades, mientras que en niños la presentación más frecuente (85%) es el edema de genitales acompañado de mayor o menor induración y eritema locales. Otro aspecto característico de la forma pediátrica es que con frecuencia aparece simultáneamente con enfermedad perianal, como en la paciente número 4. Se desconoce su fisiopatología aunque se han postulado diversos mecanismos: depósito cutáneo de inmunocomplejos circulantes, reacción de hipersensibilidad de tipo iv mediada por linfocitos, defectos en la activación de beta-2-integrinas o alteración de los mecanismos de reparación del ácido desoxirribonucleico localizados en el gen MLH-1. Puede confundirse con otras muchas dermatosis, como celulitis, erisipela, erupciones liquenoides o enfermedades de transmisión sexual. El hallazgo histológico de granulomas no caseificantes asociados a células gigantes multinucleadas tipo Langhans en la dermis superficial (preferentemente) o profunda así como en el tejido adiposo en el contexto de una EC intestinal confirma el diagnóstico. Los granulomas se localizan preferentemente en las zonas perivasculares.
Pese a que se ha descrito en ocasiones la desaparición espontánea, muchas veces el tratamiento no es satisfactorio. Diferentes fármacos han demostrado su eficacia: metronidazol, esteroides tópicos y sistémicos, azatioprina, metotrexato, ciclosporina, sulfasalacina, tetraciclinas y tacrolimus. Recientemente se ha descrito buena evolución tras tratamiento con infliximab, como es el caso de estos pacientes. Al contrario de otras MEI, la resección del intestino afectado no implica necesariamente su resolución.
ConclusionesLa EII pediátrica puede acompañarse de diferentes manifestaciones cutáneas. Pese a que en la mayoría de los casos su pronóstico es bueno y su resolución va paralela al control de la enfermedad de base, en ocasiones presentan un curso tórpido. Se ha observado buena respuesta a infliximab en estos pacientes con eritema nudoso y EC metastásica. La combinación de pioderma gangrenoso y síndrome de Sweet, excepcionalmente comunicada en la literatura médica, es un proceso de difícil tratamiento y con tendencia a dejar importantes lesiones residuales.
Autor para correspondencia.
J. Martín de Carpi
Dirección: javiermartin@hsjdbcn.org
