


Introducción
La lipomatosis encefalocraneocutánea es un cuadro poco frecuente perteneciente al grupo de los síndromes neuroectodérmicos. Descrito en 1970 por Haberland y Perou1, existen hasta el momento unos 40 descritos. Clínicamente se caracteriza por lesiones unilaterales en tejidos ectodérmicos y mesodérmicos: piel, ojo, tejido adiposo y cerebro.
Observación clínica
El paciente es el tercer hijo de padres no consanguíneos; nace a término mediante parto eutócico vaginal, con test de Apgar al minuto de 9 y a los 5 min de 10 y peso de 3.700 g. El embarazo transcurrió sin incidencias.
Al nacimiento, la exploración revela un área de alopecia de gran tamaño, de superficie lisa, en región parietotemporal derecha, así como varias lesiones papulonodulares perioculares y peribucales derechas de tamaño variable entre pocos milímetros y aproximadamente 1 cm de diámetro, una de ellas, situada en párpado, de forma polipoidea (fig. 1A). En ambos ojos se aprecian formaciones rojizas blandas en conjuntiva bulbar, que se extienden desde el canto externo del ojo hasta la región corneal, invadiéndola en el derecho (fig. 1B) y formando una excrecencia que protruye más de 1 cm por la hendidura palpebral en el izquierdo. La histología de la lesión del párpado derecho revela un pólipo fibroepitelial mientras que la de conjuntiva izquierda corresponde a un hamartoma tipo coristoma.
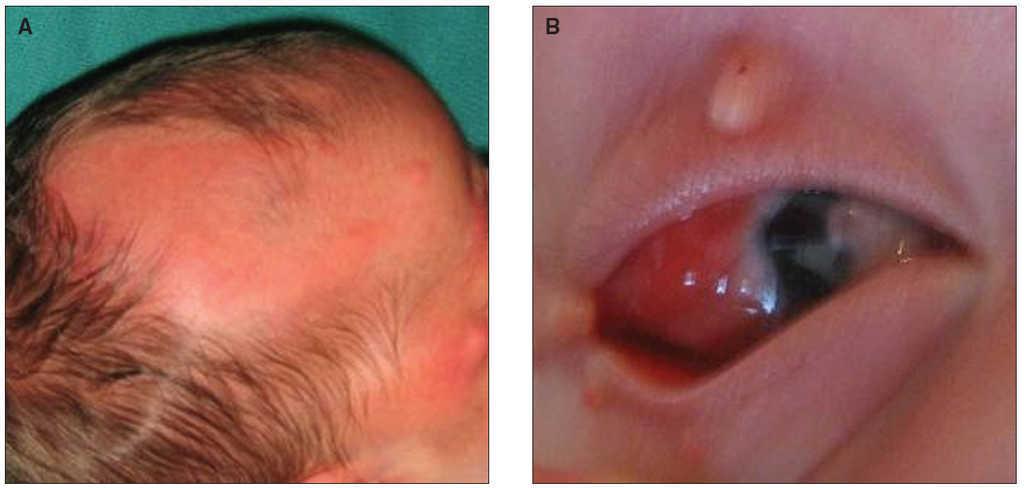
Figura 1. A) Área de alopecia en región parietotemporal derecha. B)Dermoide epibulbar que se extiende desde el canto externo invadiendo la córnea.
En la RM cerebral se observa un quiste aracnoideo de fosa temporal derecha con hipoplasia secundaria de lóbulo temporal con incompleta opercularización de la ínsula, dilatación del asta occipital del ventrículo lateral derecho acompañada de displasia cortical temporooccipital con áreas de lisencefalia y polimicrogiria (figs. 2A y 2B), pequeños lipomas a nivel de ángulo pontocerebeloso derecho y angiomas leptomeníngeos en espacio subaracnoideo occipital derecho.
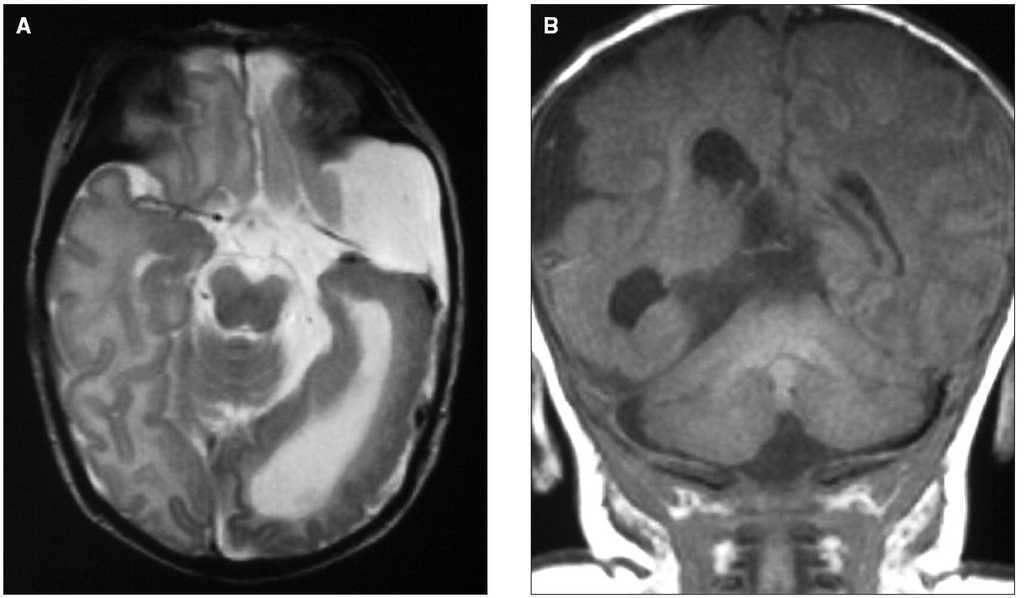
Figura 2. A)RM cerebral T2 axial; dilatación del asta occipital del ventrículo lateral derecho; displasia cortical temporooccipital con áreas de lisencefalia y polimicrogiria.B)RM cerebral T1 coronal; quiste aracnoideo de fosa temporal derecha e incompleta opercularización de la ínsula.
Se realizaron serologías TORCH, VIH y hepatitis, que fueron negativas, ecocardiograma y serie ósea completa, que fueron normales.
Se hizo el diagnóstico de lipomatosis encefalocraneocutánea o síndrome de Haberland con afectación ocular bilateral.
Discusión
El síndrome de Haberland es una disgenesia neuroectodérmica congénita de etiología desconocida. No se han demostrado anomalías cromosómicas ni ningún patrón de transmisión genética2, aunque se cree que podría ser el resultado de una mutación somática de un gen autosómico dominante que tiene lugar en una fase muy temprana de la embriogénesis y que sólo sería compatible con la vida en forma de mosaicismo2-6.
El principal marcador de la enfermedad es la presencia en región parietal o frontoparietal de un área de alopecia nítidamente demarcada, oval o redondeada, de superficie suave y a menudo cubierta por teleangiectasias, que histológicamente corresponde a una zona con ausencia de folículos pilosos y cuerdas de tejido adiposo que penetran en dermis y que recientemente se ha denominado de nevus psiloliparus4,6,7. También es típica la presencia en el mismo lado de la cara de lesiones papulonodulares normocrómicas de tamaño variable en frente, mejilla y párpado y que pueden corresponder a lipomas, fibrolipomas, hamartomas o nevos de tejido conjuntivo2,3,6-8.
La afectación ocular más típica son los coristomas o dermoides epibulbares3 ipsolaterales a las lesiones cutáneas y que pueden causar ambliopía al inducir opacidad corneal, defectos de refracción o ambos2,4.
A nivel del sistema nervioso central aparecen quistes aracnoideos en fosa craneal media, displasia o atrofia cortical con dilatación ventricular adyacente, pérdida de la normal opercularización de la ínsula y calcificaciones corticales2,3,8. También se han descrito lipomas, adelgazamiento del cuerpo calloso y angiomatosis leptomeníngea9, siempre ipsolaterales a las lesiones cutáneas. Un 60 % de los pacientes presentan epilepsia y un 40 % retraso psicomotor5, aunque no existe correlación entre la severidad de las malformaciones y las manifestaciones clínicas2.
En el caso que presentamos tanto los hallazgos clínicos como las pruebas de imagen y el estudio anatomopatológico son característicos de la lipomatosis encefalocraneocutánea, mientras que el hecho de que las lesiones oculares sean bilaterales confirman la gran heterogeneidad clínica de este síndrome y explica las dificultades que a menudo supone su diagnóstico.
Correspondencia: Dra. M. López Sousa.
Departamento de Pediatría. Hospital Clínico Universitario.
García Prieto, 15, 3.ºB. Santiago de Compostela. España.
Correo electrónico: lopezsousa@gmail.com
Recibido en abril de 2006.
Aceptado para su publicación en enero de 2007.
