



Varón, dos años, fiebre (40°C) de seis días (octubre 2020), sin cuadro catarral o vómitos. Orinas oscuras y deposiciones normales. No contacto con SARS-CoV-2, no convive con animales, no asiste a guardería, no hay antecedente de viajes recientes (originarios de Pakistán, en España desde marzo de 2020). Hijo único de pareja sana consanguínea, sin otros antecedentes. Embarazo, parto y perinatal sin incidencias. Correctamente vacunado según calendario de país de origen, este era su primer proceso febril.
Triángulo de evaluación pediátrica estable. Temperatura 39,7°C. Palidez de piel, mucosas secas, ictericia conjuntival, soplo I/VI en mesocardio, pulsos normales, hepatomegalia no dolorosa y polo de bazo palpable. Exudado amigdalar escaso y adenopatías cervicales de pequeño tamaño.
Se solicitó además de prueba de detección de antígenos (Ag) COVID-19, hemograma y bioquímica destacando afectación de serie roja con anemia (8 g/dL) microcítica e hipocrómica, neutropenia leve con leucocitos normales y trombopenia (75.000/mm3) sin alteraciones morfológicas. Test de Coombs negativo. Bilirrubina de 10 mg/dL, aumento de GPT (413 UI/L; n =1-55) y LDH (1.176 UI/L; n =125-243) y Na bajo (128 mEq/l; n =136-145). Orina normal. Se cursó estudio para virus de Epstein-Barr (VEB)/citomegalovirus (CMV); estudio para Leishmania (por endemicidad) y de gota gruesa. La prueba de Mantoux resultó de 0 mm y las pruebas de imagen mostraron hepatoesplenomegalia sin otras alteraciones.
Tras exploraciones complementarias y dada la clínica de anorexia, esplenohepatomegalia, citopenias e hipoalbuminemia en zona endémica de Leishmania, se inició tratamiento con anfotericina B liposomal. El estudio de médula ósea (MO) resultó normal sin fenómenos de hemofagocitosis ni formas sugestivas de Leishmania. Se informó resultado positivo para VEB mediante técnica PCR con resultados serológicos para Leishmania negativos, diagnosticándose de primoinfección por VEB. Quedó apirético en las primeras 24 h y descartada leishmaniasis se suspendió anfotericina B. A nivel hematológico, signos de regeneración en MO y estabilización de hemoglobina a partir de las 48 h, sin precisar transfusión.
Además de las citopenias y afectación hepática, los estudios arrojaron hiperferritinemia (7.251 ng/mL; n = 15-120) y aumento de receptor soluble de IL-2 (> 7.500 UI/L).
Por afectación hepática e hipertrigliceridemia, se descartó deficiencia de lipasa ácida lisosomal y el estudio de apolipoproteínas fue normal.
La asociación de fiebre con citopenias, aumento de ferritina, LDH, receptor soluble de IL-2, hipofibrinogenemia junto a hipertrigliceridemia, hipertransaminasemia e hiperbilirrubinemia, a pesar de una MO normal, sugirió el diagnóstico de síndrome hemofagocítico secundario a infección aguda por VEB.
Por la edad y lo excepcional de estos casos y ante consanguinidad de los padres se decidió realizar un estudio genético de síndromes hemofagocíticos primarios, confirmándose dos variantes patogénicas en heterocigosis (c.1284G>A p.(Trp428Ter) tipo nosense y c.1349C>T p.(Thr450Met) tipo missense) en el gen PRF1 (gen que codifica la perforina, proteína involucrada en la citotoxicidad de las células NK y LT citotóxicas y está relacionado con la linfohistiocitosis familiar tipo 2), y una mutación en heterocigosis de significado incierto en el gen UNC13D (c.3220C>T p.(Arg1074Trp) (gen que afecta la degranulación y produce la deficiencia de Munc 13-4 correlacionándose con el inicio de la enfermedad a los dos años o más). La citometría de flujo confirmó una expresión de perforina de 3%, citotoxicidad disminuida y degranulación normal. Se confirmó en ambos padres la presencia de una variante patogénica, aportando cada uno de ellos una mutación a su hijo. (c.1349C>T p. (Thr450Met) la madre, c.1284G>A p.(Trp428Ter) el padre).
El diagnóstico final fue de linfohistiocitosis hemofagocítica (HLH) familiar tipo 2, y tras revisión de literatura, se remitió a centro de referencia para búsqueda de donante y realización de trasplante de progenitores hematopoyéticos, estando el paciente actualmente estable.
Los hallazgos clínicos y de laboratorio se relacionan con la fisiopatología de la HLH1. La fiebre es resultado de niveles altos de IL-6. La esplenomegalia, de la infiltración de linfocitos y macrófagos y las citopenias por concentraciones elevadas de factor de necrosis tumoral (TNF) -α e interferón (IFN) -γ, así como por hemofagocitosis directa. La hipertrigliceridemia se atribuye a la disminución de la actividad de la lipoproteína lipasa iniciada por el aumento de los niveles de TNF-α y los linfocitos activados producen altas concentraciones de receptor de IL-2 soluble2,3.
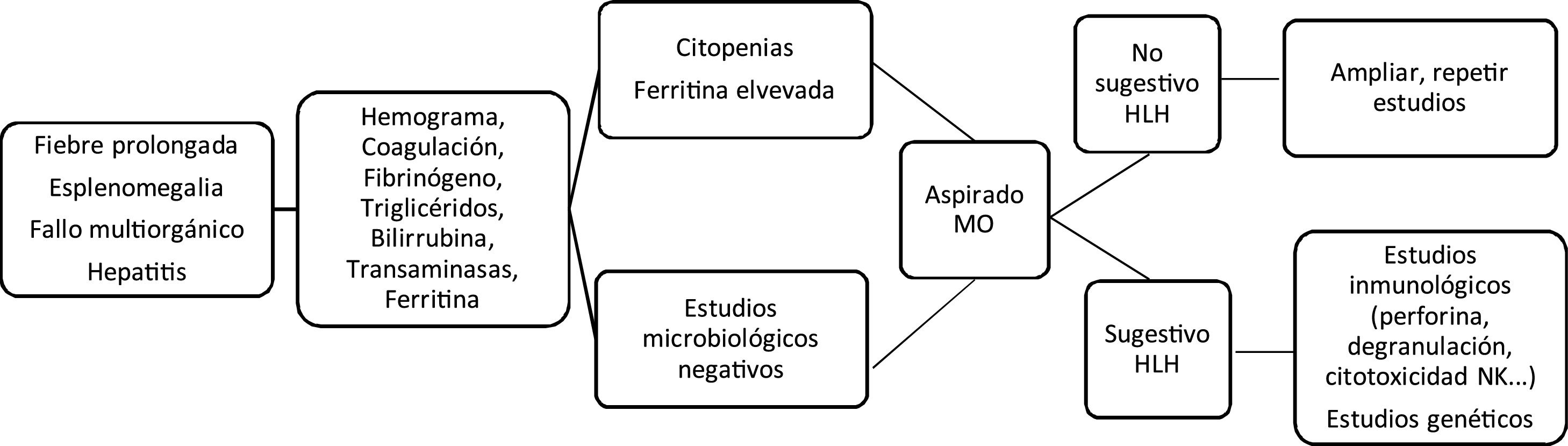
Los signos clínicos iniciales recuerdan cualquier proceso infeccioso grave. Fiebre alta prolongada, y de forma progresiva pancitopenia y hepatoesplenomegalia, junto con datos de fallo multiorgánico. En el laboratorio las citopenias, la coagulopatía con hipofibrinogenemia, la hipertrigliceridemia, hipertransaminasemia e hiperferritinemia apoyan la sospecha diagnóstica (fig. 1). Estos eventos conforman los criterios clínicos y de laboratorio utilizados para el diagnóstico, establecidos en 1991 y revisados en 2004. Por esto debe ser incluido en el diagnóstico diferencial de fiebre de origen desconocido, fallo hepático agudo, hepatitis con coagulopatía o sepsis con fallo multiorgánico3,4.
Las diferentes modalidades terapéuticas incluyen corticoides, inmunosupresores, citostáticos, inmunomoduladores, fármacos biológicos o anticuerpos monoclonales, según sea el caso, pero en formas genéticas, el tratamiento curativo es el trasplante de progenitores hematopoyéticos para corregir el defecto de citotoxicidad5,6.
Hay formas clínicas muy graves de progresión fulminante que requieren terapia específica y medidas de soporte intensivo de forma empírica y urgente, mientras que otros casos son más larvados o cursan de forma recidivante y responden a tratamientos de soporte menos agresivos hasta poder realizar el tratamiento definitivo como ocurrió en nuestro caso.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
