



A boy aged 2 years presented with fever of 6 days’ duration (40 °C) in October 2020, without symptoms of upper respiratory tract infection or vomiting. The urine was dark and the stools were normal. The patient had not been exposed to SARS-CoV-2, there were no animals in the household and he had no history of recent travel (the family was from Pakistan and had been in Spain since March 2020). He was the single child of a healthy consanguineous couple, with no other history of interest. There had been no complications during pregnancy, delivery or the perinatal period. The patient was correctly vaccinated according to the Pakistani vaccination schedule, and this was his first episode of febrile illness.
The paediatric assessment triangle indicated the patient was stable. His temperature was 39.7°C. Other symptoms included pallor, mucosal dryness, conjunctival icterus, a murmur grade I/VI at the level of the mesocardium, normal pulses, painless hepatomegaly and palpable spleen tip. There was a small amount of tonsillar exudate and mild cervical lymph node enlargement.
In addition to the SARS-CoV-2 antibody test, a complete blood count with differential and blood chemistry panel were ordered, of which the salient findings were microcytic hypochromic anaemia (red blood cells, 8g/dL), mild neutropaenia with a normal white blood cell count and thrombopenia (75 000 platelets/mm3) without morphological abnormalities. The Coombs test was negative. The bilirubin level was 10mg/dL, with elevation of alanine aminotransferase (ALT, 413 IU/L; normal range: 1–55) and lactate dehydrogenase (LDH, 1176 IU/L; normal: 125–243) and decreased sodium (Na, 128 mEq/L; normal: 136–145). The urinalysis was normal. The evaluation also included an Epstein-Barr virus (EBV)/cytomegalovirus (CMV) test; testing for leishmaniasis (endemic country of origin) and a thick blood film examination. The Mantoux test reaction measured 0mm and imaging tests evinced hepatosplenomegaly in the absence of other abnormalities.
After performing the initial tests and given the presentation with decreased appetite, hepatosplenomegaly, cytopenia and hypoalbuminaemia in a patient from a region with a high burden of leishmaniasis, treatment was initiated with liposomal amphotericin B. The bone marrow (BM) examination was normal, with no evidence of haemophagocytosis or morphology suggestive of leishmaniasis. The EBV polymerase chain reaction (PCR) test turned out positive, while serological tests for detection of Leishmania were negative, leading to diagnosis of primary infection by EBV. The patient became afebrile within 24h and, having ruled out leishmaniasis, treatment with amphotericin B was discontinued. At the haematological level, there were signs of BM regeneration and stabilization of haemoglobin levels from 48h, and the patient did not require transfusion.
In addition to the cytopenias and liver involvement, the evaluation evinced Hyperferritinaemia (ferritin, 7251ng/mL; normal: 15–120) and increased levels of the soluble interleukin (IL)-2 receptor (>7500 IU/L).
On account of the hepatic involvement and hypertriglyceridemia, lysosomal acid lipase deficiency was ruled out, and apolipoprotein test results were normal.
The presence of fever associated with cytopenias, elevation of ferritin, LDH and soluble IL-2 receptor, hypofibrinogenaemia combined with hypertriglyceridemia, hypertransaminasaemia and hyperbilirubinaemia, despite a normal BM, suggested the diagnosis of haemophagocytic syndrome secondary to acute infection by EBV.
Given the age of the patient, the rarity of the presentation and the consanguinity of the parents, genetic testing was performed for screening of primary haemophagocytic syndromes, confirming the presence of two pathogenic heterozygous variants—nonsense variant c.1284G>A p.(Trp428Ter) and missense variant c.1349C>T p.(Thr450Met)—in gene PRF1 (which encodes perforin, a protein involved in the function of cytotoxic NK and T cells and associated with familial haemophagocytic lymphohistiocytosis type 2) and a heterozygous variant of uncertain significance—c. 3220C>T p.(Arg1074Tr)—in gene UNC13D (a variant that affects degranulation and causes Munc13-4 deficiency, which is associated with the onset of the disease from age 2 years). Flow cytometric analysis confirmed a perforin expression of 3%, impaired cytotoxicity and normal degranulation. The presence of a pathogenic variant was confirmed in both parents, each of whom contributed one variant to the child—c. 1349C>T p. (Thr450Met) in the mother, and c.1284G>A p.(Trp428Ter) in the father.
The final diagnosis was familial haemophagocytic lymphohistiocytosis (FHL) type 2, and, following a review of the literature, the patient, stable at the time, was referred to a reference centre to search for a donor and performance of haematopoietic stem cell transplantation.
The clinical and laboratory features were consistent with the pathophysiology of FHL.1 Fever results from the increased levels of IL-6. Splenomegaly, lymphocyte and macrocyte infiltration and cytopenias result from elevated levels of tumour necrosis factor α (TNF-α), and interferon γ (IFN-γ) as well as direct haemophagocytosis. Hypertriglyceridemia is attributed to the reduction in lipoprotein lipase activity resulting from the elevation of TNF-α levels and activated lymphocytes produce high concentrations of soluble IL-2 receptor.2,3
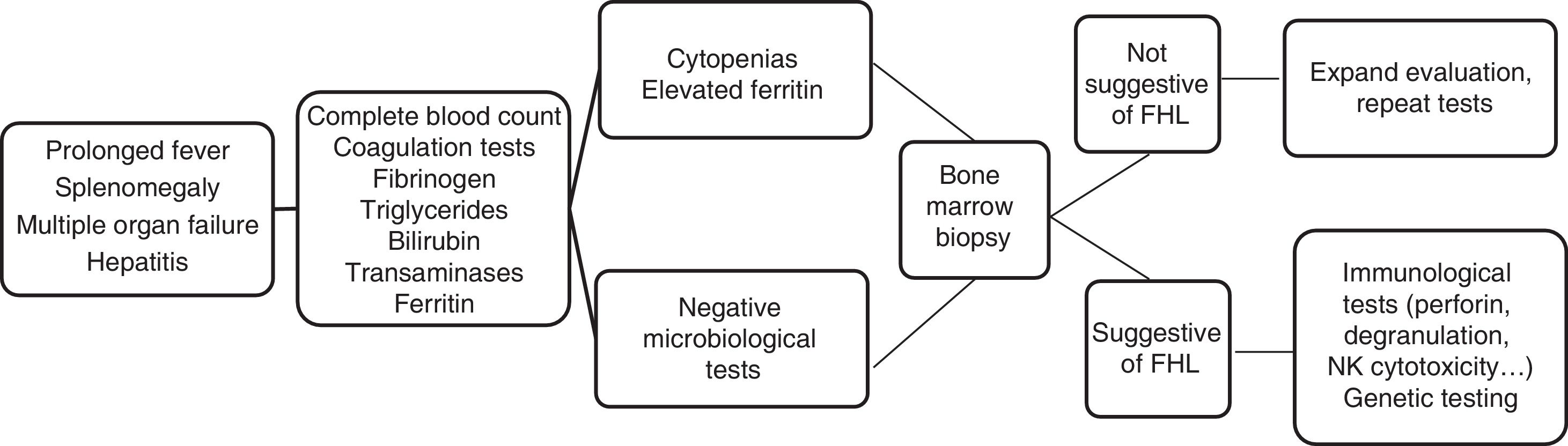
The initial clinical manifestations were similar to those found in any severe infection. There was a prolonged high fever and progressive pancytopenia and hepatosplenomegaly associated with features suggestive of multiple organ failure. In the complete blood count and chemistry panel, the presence of cytopenias, coagulopathy with hypofibrinogenaemia, hypertriglyceridemia, hypertransaminasaemia and hyperferritinaemia supported the suspected diagnosis (Fig. 1). These findings correspond to the clinical and laboratory criteria required for diagnosis established in 1991 and updated in 2004. Therefore, FHL should be included in the differential diagnosis of patients with fever of unknown source, acute liver failure, hepatitis with coagulopathy or sepsis with multiple organ failure.3,4
The treatment options include steroids, immunosuppressants, cytostatic agents, immunomodulators, biological agents or monoclonal antibodies, depending on the case, but in the case of a genetic aetiology, haematopoietic stem cell transplantation is the curative treatment used to correct the impairment in cytotoxic function.5,6
There are very severe forms of disease with fulminant progression that require specific treatment and intensive supportive care on an empiric and urgent basis, while other forms develop more slowly or have a recurrent course and respond to less aggressive supportive care while awaiting definitive treatment, as was the case of our patient.
Conflicts of interestThe authors have no conflicts of interest to declare.
