


La linfangiectasia intestinal (LI) es una causa inusual de enteropatía pierde-proteínas (EPP)1 y puede ser primaria (LIP) o secundaria (LIS). La LIP, que describió Waldmann en 1961, es una linfopatía intestinal congénita caracterizada por la dilatación de los vasos linfáticos y el paso de linfa hacia la luz intestinal, con pérdida crónica de proteínas, grasa, linfocitos e inmunoglobulinas por las heces. La obstrucción linfática puede ser secundaria a origen cardíaco (postintervención de Fontan)2 o debida a procesos inflamatorios intestinales3,4 (neoplasias abdominales o retroperitoneales, posquimioterapia o radiación, tuberculosis mesentérica, etc.). Presentamos 2 casos de LIP que se manifiestan con diferente espectro clínico y edad de inicio.
Caso 1. Niño con inicio a los 2 años con edemas generalizados, oliguria, astenia, anorexia y heces blandas. Antecedentes personales: linfedema congénito y meningitis neumocócica a los 6 meses de edad.
Peso: 16 kg (percentil [P] 97), talla: 87 cm (P50). Edemas generalizados, ascitis y linfedema de extremidades superiores. En la analítica se observa hipoproteinemia (3,7 g/l), hipoalbuminemia (2,4 g/dl), hipocalcemia y linfocitopenia. Hipocolesterolemia, hipotrigliceridemia e hipogammaglobulinemia (inmunoglobulina [Ig] G: 92 mg/dl, IgA: 24 mg/dl e IgM: 47 mg/dl). Linfocitos T (LT) CD4: 10%; LT CD8: 8% con inversión CD4/CD8. En la orina de 24 h se descarta proteinuria. La alfa-1-antitripsina (α1-AT) en heces es de 5,2 mg/g (valor normal [VN]: inferior a 0,3 mg/g). El tránsito esofagogastroduodenal (TEGD) muestra pliegues mucosos de intestino delgado engrosados. La tomografía computarizada (TC) abdominal muestra edema mesentérico difuso. En la fibroendoscopia gastroduodenal (FEGD) se observa mucosa duodenal de aspecto lechoso e imágenes «en copos de nieve». La anatomía patológica (AP) muestra marcada dilatación de vasos linfáticos, patognomónica de LI. El tratamiento fue sustitutivo con seroalbúmina y gammaglobulinas, dieta hiperproteica e hipograsa (enriquecida con triglicéridos de cadena media).
Actualmente, a los 10 años el niño tiene un correcto desarrollo pondoestatural y sin infecciones graves, a pesar de que mantiene un patrón bioquímico de pérdida linfática con hipogammaglobulinemia.
Caso 2. Niña con inicio a los 6 años en forma de edema bipalpebral, deposiciones dispépticas y epigastralgia. Antecedentes: varicela a los 2 años y herpes zóster un mes antes del inicio); tía materna con tuberculosis pulmonar en el año previo. Peso: 21,2 kg (P50), talla: 116 cm (P50–75). Ojerosa, con palidez cutánea. Discreto edema bipalpebral.
En la analítica se observa hipoproteinemia (2,5 g/dl), hipoalbuminemia (1 g/dl), hipocalcemia y linfocitopenia. Hipocolesterolemia, hipotrigliceridemia e hipogammaglobulinemia (IgG: <160 mg/dl; IgA: 36 mg/dl; IgM: 51 mg/dl). LT CD4 12%, LT CD8 34% con inversión CD4/CD8. El estudio serológico dio resultado negativo, salvo para el virus de la varicela-zóster (VVZ) (IgG: 4,12 [VN<1,0]; IgM: 0,81 [VN<1.1]). En la orina se descarta proteinuria; la reacción en cadena de la polimerasa para citomegalovirus resulta negativa. La α1-AT en heces es de 2,48 mg/g. Coprocultivo, parásitos en heces, marcadores de celiaquía, test radioalergosorbente a proteínas de leche de vaca, test de Mantoux y las técnicas QuantiFERON® y TB-Spot resultan todos negativos. La radiografía de tórax, la ecocardiografía y el TEGD fueron normales. En la TC abdominal se visualizan múltiples adenopatías retroperitoneales de pequeño tamaño. La FEGD muestra mucosa duodenal de aspecto lechoso e imágenes «en copos de nieve» (fig. 1). En la AP se observa mínima dilatación de vasos linfáticos en porción apical de una vellosidad. El tratamiento es sustitutivo y dietético, asociado a profilaxis para Pneumocystis jirovecii. Se deriva a la niña a un centro de tercer nivel para completar el estudio. La videocápsula endoscópica realizada a los 3 meses de iniciado el tratamiento (paciente asintomática, analítica normal y α1-AT en heces: 0,12 mg/g) no muestra alteración. Con la orientación diagnóstica de LIS a infección por VVZ se inicia reintroducción progresiva de dieta normal con buena tolerancia, aunque con persistencia de hipogammaglobulinemia moderada. A los pocos meses presenta empeoramiento progresivo con reaparición de los edemas palpebrales, albúmina de 1,4 g/dl, IgG de 230 mg/dl y α1-AT en heces de 2,05 mg/g. Una nueva FEGD con imágenes indicativas y AP revisada compatible confirman el diagnóstico de LI. Evolución satisfactoria al readministrar el tratamiento dietético, sin necesidad actual de tratamiento sustitutivo.
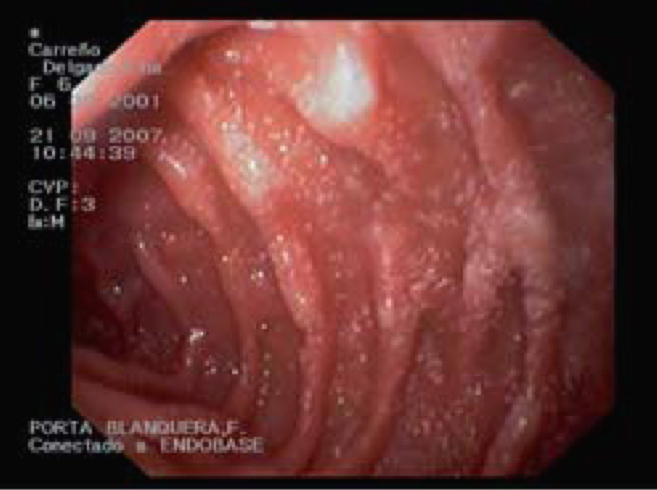
Figura 1. Fibroendoscopia gastroduodenal en caso clínico 2: imagen en «copos de nieve» característica de linfangiectasia intestinal.
La EPP puede tener un origen multifactorial y plantea un diagnóstico diferencial extenso. Como muestran nuestros 2 pacientes, la LI puede presentarse con un amplio espectro clínico y requiere un alto índice de sospecha en los casos con manifestaciones más leves. Su diagnóstico es dificultoso cuando las dilataciones linfáticas no son demostrables histológicamente por ser focales. La videocápsula endoscópica ha demostrado ser útil para valorar la localización y extensión de la LI, especialmente cuando ésta afecta a tramos no accesibles a la FEGD5,6.
El diagnóstico en el primer caso clínico fue sencillo; la clínica y las dilataciones linfáticas patognomónicas lo confirmaron. El segundo caso precisó una mayor serie de pruebas, debido a que las muestras histológicas se informaron como inespecíficas, a pesar de observarse mínimas dilataciones linfáticas. Inicialmente se planteó como diagnóstico más probable una LIS a infección por VVZ. Hay escasos casos publicados de varicela y EPP secundaria a la afectación del tracto gastrointestinal por VVZ7. Ross et al8 publicaron el caso de una mujer de 46 años con varicela grave y LIS, pero no hemos encontrado ningún caso pediátrico. Después de la segunda FEGD, ante la firme sospecha de LIP se revisa la histología, en la que se aprecian (de forma sutil) dilataciones linfáticas en la parte apical de la vellosidad y enterocitos cargados de grasa en las últimas muestras (fig. 2).
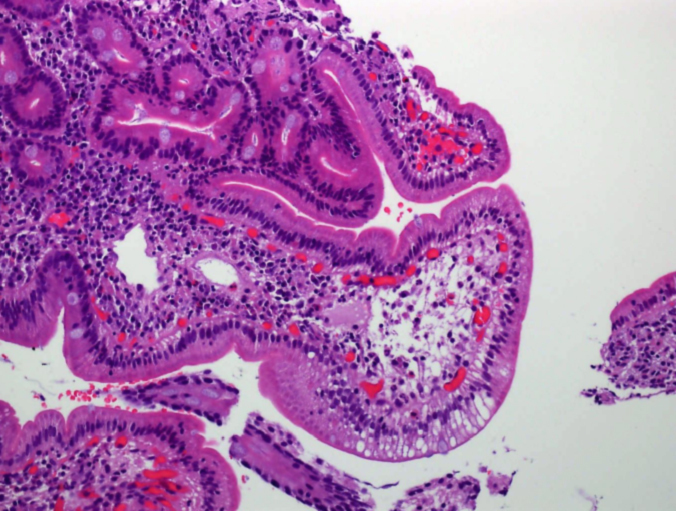
Figura 2. Anatomía patológica en caso clínico 2: dilataciones linfáticas en punta de vellosidad con enterocitos cargados de material lipídico.
La LIP es un ejemplo típico de inmunodeficiencia secundaria. Las concentraciones de inmunoglobulinas están disminuidas pero su producción es normal; la inmunidad celular también está afectada por la linfocitopenia mantenida. A pesar de esto, en los pacientes con LIP se desarrollan infecciones graves o recurrentes con poca frecuencia9,10.
Autor para correspondencia.
Z. Lobato Salinas
Dirección: zulosa@gmail.com
