



Caso clínico
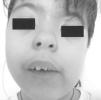
Niña de 10 años remitida por sangrado vaginal de 3 días de duración. Padres sanos, hija única. Sin abortos ni mortinatos. Plagiocefalia por cierre unilateral de sutura lamboidea a los 18 meses. No toma medicamentos. Conducta autista. Recibe educación especial. Desarrollo mamario a los 10 1/2 años. Exploración: 40 kg (+1,20 DE), 138,9 cm (+0,38 DE), PA 95/45 mmHg, Tanner: S3P3, edad ósea 11,5 años. Epicanto bilateral, puente nasal ancho, sinefridia, paladar ojival, boca entreabierta continua, orejas de implantación baja, retrognatia, asimetría facial, estrabismo (fig. 1). Manos y pies normales. Plagiocefalia (fig. 2). A los 3 meses aprecian lesiones cutáneas hipopigmentadas, parcheadas, generalizadas en tronco, espalda y extremidades (fig. 3). Exámenes complementarios: coxa valga bilateral sin escoliosis ni dismetrías; polo anterior y fondo ocular normales. EEG y RM: normales. Hormonas tiroideas y cortisol normales. FSH: 7,6 U/l, LH: 3,5 U/l. Cariotipo periférico 46XX. Prueba de GnRH: pico de LH 30 U/l y de FSH 18 U/l, estradiol: 100 pg/ml. IGF-I: 469 ng/ml). Ecografía: útero de 4 × 3,9 × 1,7 cm, ovario derecho de 3,2 ml (con folículos de 7 mm) y ovario izquierdo de 0,7 ml. Ante el retraso psicomotor y para mejorar su adaptación social se trató con triptorelina 3,75 mg/28 días que se retiró a los 12,5 años de edad ósea, tras 8 dosis. Ante la sospecha clínica, se realizó prueba diagnóstica.
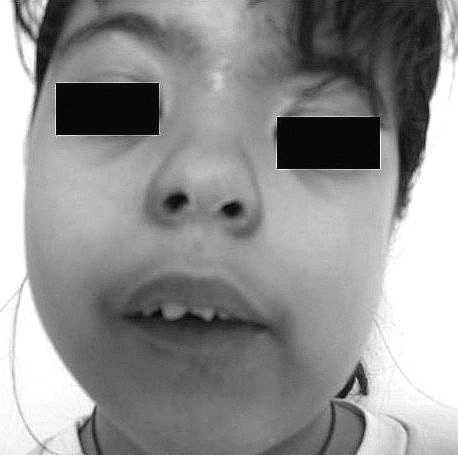
Figura 1. Rostro de la paciente.
Figura 2.RM: plagiocefalia.

Figura 3. Lesiones cutáneas hipopigmentadas.
Pregunta
¿Cuál es su diagnóstico?
Mosaicismo pigmentario tipo Ito (hipomelanosis de Ito)
Se realizó cariotipo de fibroblastos de una lesión hipopigmentada sin apreciarse anomalías en las bandas G.
A pesar de no haber detectado alteraciones cromosómicas el diagnóstico1,2 se basa en un criterio sine qua non (manchas hipopigmentadas lineales o parcheadas congénitas no hereditarias) más alteraciones neurológicas y musculoesqueléticas. La presencia de anomalías cromosómicas es un criterio menor.
Fue descrita en 1951 por Ito y bautizada como hipomelanosis de Ito (HI) por Jelinek en 19733,4. Abarca un grupo heterogéneo de alteraciones con presencia de remolinos, manchas y líneas hipopigmentadas bien delimitadas que siguen las líneas de Blaschko (sin afectar palmas y plantas) junto con déficit neurológicos, epilepsia y alteraciones asimétricas en otros órganos4-6. El 54 % tienen hipopigmentación al nacimiento y el 70 % antes del año, que no cambia en la infancia pero va desapareciendo en la vida adulta5.
Es la tercera neurodermatosis en frecuencia5,7 y se presenta en 1/7.895 consultas dermatológicas, 1/2.893 consultas de pediatría general4 y 1/700 consultas de neuropediatría7. Pasa desapercibida frecuentemente por inadvertencia de las lesiones cutáneas y su trascendencia1,4,7.
Entre las alteraciones extracutáneas las neurológicas son las más prevalentes (76 %), seguidas de las osteomusculares, faciales y oculares. Son menos frecuentes las cardíacas, urogenitales y la pubertad precoz1,5. Las alteraciones asociadas se describen más por los neurólogos (94 %) que por los dermatólogos (66 %)8. Los tumores son infrecuentes y suelen aparecer en los primeros años de vida8.
Las líneas de Blaschko no siguen ninguna estructura anatómica nerviosa, vascular o linfática. Dibujan clones celulares de las fases tempranas del desarrollo ectodérmico y reflejan un mosaicismo germinal5,6.
El análisis citogenético revela frecuentemente mosaicismo cromosómico con una gran variedad de anomalías (poliploidía, aneuploidía, deleciones cromosómicas, inserciones, translocaciones) que pueden afectar a cualquier par de cromosomas, lo que explicaría la heterogeneidad de las manifestaciones clínicas en la HI6.
Estas alteraciones cromosómicas se expresan como un mosaicismo pigmentario (MP). En una serie de de 113 casos con anomalías citogenéticas sólo un 30-60 % mostraron alteraciones en el cariotipo6 probablemente porque la anomalía sólo se encuentra en las lesiones cutáneas e incluso es posible que sólo se encuentre en los derivados ectodérmicos (queratinocitos y melanocitos) y no en los fibroblastos que son los únicos que suelen cultivarse3,6. En el 80 % de los MP la anomalía cromosómica coincidía con regiones en las que existen genes que intervienen en la pigmentación cutánea y en la diferenciación de las células de la cresta neural y en la migración de los melanoblastos.
En la HI con lesiones despigmentadas los melanocitos estarían ausentes y en las lesiones hipopigmentadas éstos serían parcialmente funcionales6. Para avanzar en el campo del MP es preciso optimizar las técnicas citogenéticas utilizando queratinocitos o melanocitos más bien que linfocitos o fibroblastos para detectar microdeleciones6. Las anomalías cromosómicas no aumentan el riesgo de retraso psicomotor pero sí el de tumores cerebrales.
La HI es esporádica y en menos del 3 % de familias tiene herencia autosómica dominante, siendo el consejo genético obligatorio.
La histología óptica y electrónica de la IH es inespecífica pero útil para el diagnóstico diferencial y se han descrito cambios en los melanocitos y premelanosomas4.
La HI no es una entidad única y sería una manifestación inespecífica (fenotipo) de diferentes mosaicismos. Es un término descriptivo, y se aplica a pacientes con patrones de despigmentación o hipopigmentación difusos, parcheados, generalizados, limitados o punteados, distribuidos en las líneas de Blaschko. Los pacientes con despigmentación, hiperpigmentación o hipopigmentación y sin diagnóstico específico (piebaldismo, síndrome de Waardenburg o IP) deben ser sometidos a un cariotipo3, y dado que existen pacientes con patrones cutáneos variados siguiendo las líneas de Blaschko con un mosaicismo demostrado deben ser diagnosticados de HI4.
Por todo ello, el término HI debería eliminarse y sustituirse por displasia pigmentaria, mosaicismo pigmentario tipo Ito o hipopigmentación a lo largo de las líneas de Blaschko, siendo preferible el de MP o MP tipo Ito4,8.
Agradecimiento
A la Dra. Roselló, del Servicio de Genética y Diagnóstico Prenatal del Hospital Universitario La Fe de Valencia por el cariotipo de la biopsia cutánea.
Correspondencia: Dr. F. Aleixandre Blanquer.
Servicio de Pediatría. Hospital General de Área Virgen de la Salud.
Camino Colonia Romana, 12, 9.º A. 03016 Alicante. España.
Correo electrónico: faleixandre@gmail.com
Recibido en mayo de 2006.
Aceptado para su publicación en julio de 2006.
