


En relación con el interesante artículo «Lesiones autoinvolutivas en histiocitosis de células de Langerhans» de los autores Tous-Romero et al., publicado en Anales de Pediatría el pasado mes de diciembre1, creemos necesario insistir en algunos aspectos referentes a esta patología.
La histiocitosis de células de Langerhans constituye un grupo heterogéneo de enfermedades, que abarca desde la afectación cutánea aislada hasta una enfermedad multisistémica grave2. Consiste en una proliferación clonal de células dendríticas de origen mieloide con etiología discutida3. La afectación cutánea congénita aislada es infrecuente y suele tener buen pronóstico. La denominación «histiocitosis congénita autoinvolutiva», empleada por los autores del mencionado artículo, actualmente no se considera adecuada4 por la documentación de casos de histiocitosis neonatal congénita de resolución espontánea que han evolucionado a enfermedad multisistémica tras la desaparición de las lesiones cutáneas (del 8-13% según las series5,6). Es posible que la frecuencia real de la forma cutánea aislada esté infraestimada, debido al polimorfismo, escasez en número, e inespecificidad de las lesiones cutáneas, como reflejan los autores en su publicación y reiteramos a continuación.
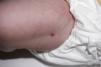
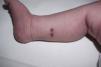
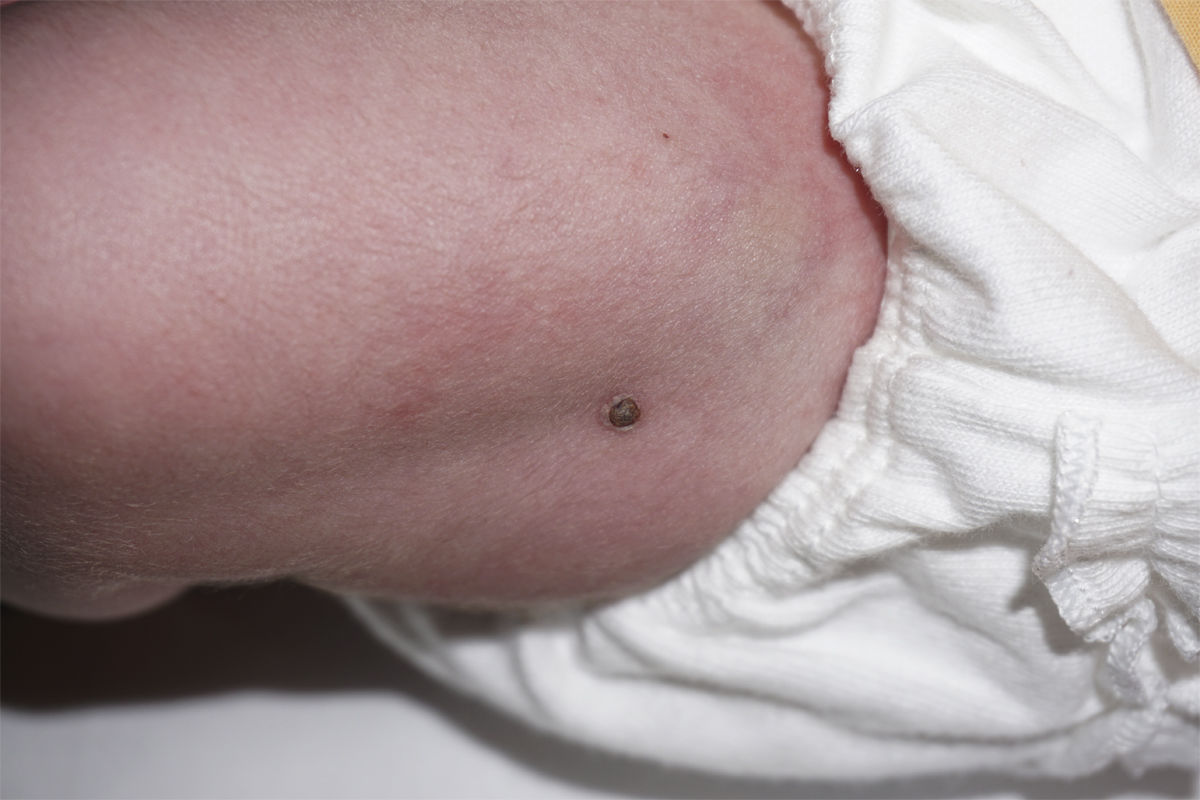
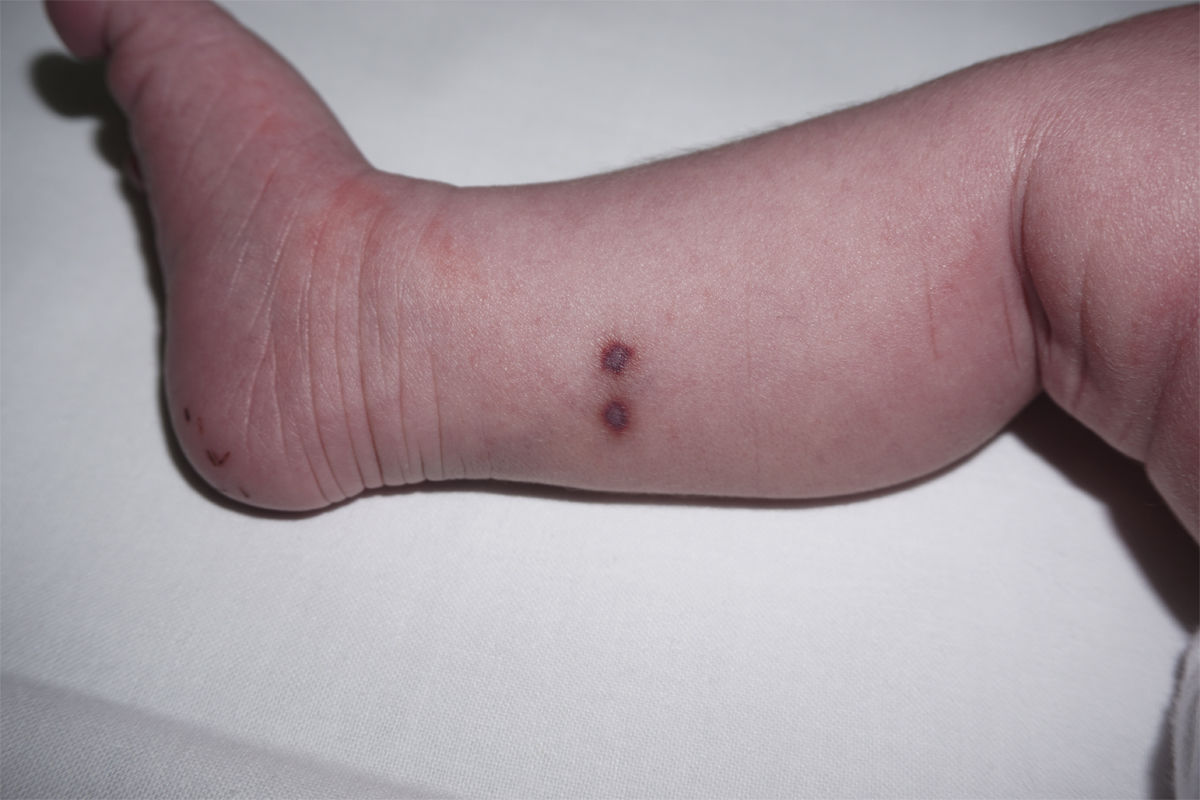
Presentamos el caso de una recién nacida a término sin antecedentes de interés ingresada en Neonatología por hipoglucemia precoz asintomática con aportes intravenosos de glucosa durante 48 h. El primer día de vida, se objetivó una lesión nodular sobreelevada de 4mm en muslo izquierdo, costrosa con collarete descamativo (fig. 1). Se planteó duda diagnóstica con una lesión secundaria a la administración intramuscular de vitamina K. Al tercer día de vida, se evidenciaron dos nuevas lesiones purpúricas infiltradas en el tobillo derecho, de 3-4mm de diámetro (fig. 2). Se sugirió diagnóstico diferencial con lesión secundaria a extravasación de un acceso vascular, pero, ante el aspecto infiltrado, se realizó interconsulta a Dermatología, que biopsió las lesiones. La histopatología mostró células con abundante citoplasma y núcleo lobulado que asemejan células de Langerhans, sobre fondo inflamatorio. La inmunohistoquímica fue positiva para CD1a y CD207. Se realizó estudio de extensión con analítica sanguínea, ecografía abdominal y serie ósea que resultó normal y seguimiento clínico y analítico al mes, 2 y 6 meses con resolución de las lesiones a los 2 meses de vida. No se realizó estudio de médula ósea por decisión paterna. Se diagnosticó de histiocitosis de Langerhans congénita cutánea sin afectación sistémica. La paciente continúa asintomática al año de edad.
La gran variedad de lesiones cutáneas y su tendencia a la resolución espontánea en semanas, podría contribuir a la falta de diagnóstico de una enfermedad que precisa, según recomendaciones internacionales2, seguimiento por su posible progresión multisistémica. Los porcentajes publicados de progresión de enfermedad pueden no ser exactos; es posible que formas de comienzo multisistémico tengan antecedentes de lesión cutánea inadvertida o los pacientes con enfermedad limitada a la piel presenten estudio de extensión incompleto5.
Creemos fundamental considerar esta entidad en el diagnóstico diferencial de lesiones cutáneas neonatales, en muchas ocasiones realizado por pediatras y no dermatólogos, por la importancia de realizar diagnóstico de extensión5 y seguimiento clínico estrecho.
Al Servicio de Dermatología Pediátrica, Neonatología y Oncohematología Pediátrica del Hospital General Universitario Gregorio Marañón.
