


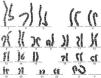
El síndrome de Turner se diagnostica mediante la combinación de ciertas características fenotípicas con la ausencia de un cromosoma X. Esta ausencia puede ser total o parcial, como la que tiene lugar en los isocromosomas X. Las consecuencias fenotípicas de estos dependen de 2 factores: la naturaleza de los genes que se han perdido y el porcentaje de células 45, X en los mosaicismos.
La clínica cambia en función del patrón citogenético, prevaleciendo la talla baja como manifestación fenotípica, ya que radica en la haploinsuficiencia del gen SHOX en el brazo corto del cromosoma X. Así, cuando existen isocromosomas de los brazos largos, la talla baja siempre está presente. Sin embargo, los rasgos típicos pueden estar ausentes, conllevando retraso diagnóstico. Este hecho se da en nuestras pacientes, y a causa de ello se beneficiarán en menor medida del tratamiento con GH.
Turner syndrome is diagnosed by the combination of certain phenotypic characteristics with the absence of one of the X chromosome. This absence may be total or partial, as occurs in isochromosomes Xq. The phenotypic consequences of these depend on two factors: the characteristics of the lost genes and the percentage of cells 45, X in mosaicisms.
The clinical features also change with the cytogenetic pattern. Short stature is the most common phenotypic manifestation, as it is due to the haploinsufficiency of the SHOX gene on the short arm of X chromosomes. Thus, when there is isochromosomes on the long arms, short stature is always present. However, the typical features of this syndrome could be absent, and the diagnosis can be delayed. This occurred in our patients, who will not be able to obtain optimum benefits with growth hormone treatment.
El síndrome de Turner se diagnostica mediante la combinación de ciertas características fenotípicas con la ausencia de un cromosoma X, que puede ser parcial como la que tiene lugar en los isocromosomas X. Es la isocromosomopatía más frecuente en el ser humano, por lo que el cariotipo en sangre periférica debe ser uno de los primeros pasos en el diagnóstico.
La clínica puede variar en función del patrón citogenético, prevaleciendo la talla baja como manifestación fenotípica más frecuente, normalmente con desproporción entre el hemicuerpo superior e inferior. Otras son las manifestaciones derivadas del hipogonadismo primario, que aparecen en la etapa puberal. Sin embargo rasgos típicos pueden estar ausentes, conllevando en ocasiones un retraso en el diagnóstico.
Presentamos 2 casos de diagnóstico tardío de síndrome de Turner dadas las escasas manifestaciones fenotípicas, ambos con diagnóstico citogenético de isocromosoma X.
Caso 1Niña de 10 años y 5 meses derivada por su médico de zona a consultas de endocrinología pediátrica para valoración de talla baja e inicio de botón mamario bilateral. Es nacida en España, hija de padre español y madre ecuatoriana, con talla genética final de 163,2cm±5cm (p50, DE: –0,11 Carrascosa et al., 2010). Presenta estancamiento ponderal desde los 8 años, con crecimiento progresivo pero enlentecido desde los 3 años.
Presentó un peso al nacer de 2.700g (p90, DE: 1,16), y talla 47cm (p75, DE: 0,79) a la edad gestacional de 35 semanas, con oligoamnios aunque sin otras complicaciones perinatales. Presenta un desarrollo psicomotor normal, sin retraso mental.
En consulta de endocrinología se determina, a una edad de 10 años y 5 meses, talla de 124,8cm (p<1, DE: –2,69) y peso de 33,7kg (p25, DE: –0,55), con velocidad de crecimiento en el último año de 3cm/año (p<1, DE: –2,51 Ferrández et al.1). Presenta fenotipo normal, con Tanner 2, telarquia 2, pubarquia 1 y sin axilarquia.
Los resultados de las pruebas complementarias muestran discreta policitemia: hemoglobina de 15,2g/dl, hematíes 5,34×10e6μl, y hematocrito 46,2% con volumen corpuscular normal (86,4fl). El estudio bioquímico de función hepatorrenal, glucídico o lipídico no muestra alteraciones. El estudio hormonal evidencia función tiroidea normal (T4L: 1,30ng/dl, TSH: 2,2mUI/ml), y patrón de gonadotropinas con LH de 6,3mUI/ml, FSH de 33,8mUI/ml y estradiol de 22pg/ml. Asimismo se solicita GH basal de 0,6ng/ml con pico tras estimulación con clonidina de 10,1ng/dl y un valor de IGF-1 de 156,5ng/ml (>p10). La edad ósea no muestra alteraciones respecto a la edad cronológica (10 años).
Se solicita cariotipo (fig. 1), donde en todas las metafases estudiadas se observan 46 cromosomas, objetivándose la presencia de una alteración estructural consistente en un isocromosoma de los brazos largos de uno de los cromosomas X, siendo una variante de síndrome de Turner.
El resto de exámenes complementarios realizados descartan malformaciones renales (ecografía abdominal normal), alteraciones neurohipofisarias (resonancia magnética sin alteraciones) y enfermedad celíaca (antitransglutaminasa tisular y antigliadina negativos). El ecocardiograma realizado muestra una válvula bicúspide aórtica con resto del estudio normal. Una vez diagnosticada, se inicia tratamiento con GH en dosis de 1,5mg/día.
Caso 2Niña de 9 años y medio en seguimiento en consultas de neurología pediátrica desde los 7 años por presentar hipoplasia de vermis cerebeloso y nistagmo horizontal. Talla genética de 171,5cm±5cm (p89, DE: 1,27). Presenta velocidad de crecimiento en p5-p10.
Es una recién nacida a término con un peso de 2.720g (p11, DE: –1,25), talla de 49cm (p40, DE: −0,26), perímetro craneal de 33cm (p20, DE: −0,86). Presenta desarrollo psicomotor y rendimiento escolar normales, sin otros antecedentes de interés.
La medición en endocrinología muestra una talla de 125,4cm (p3, DE: −1,87), con peso de 24,6kg (p10, DE: −1,32). Presenta un fenotipo en el que destaca la presencia de paladar hendido y acortamiento del 4.° metacarpiano bilateral. Se observan, además, nevus melanocíticos en tronco. El resto de la exploración no muestra hallazgos relevantes y el estudio cardiológico fue normal.
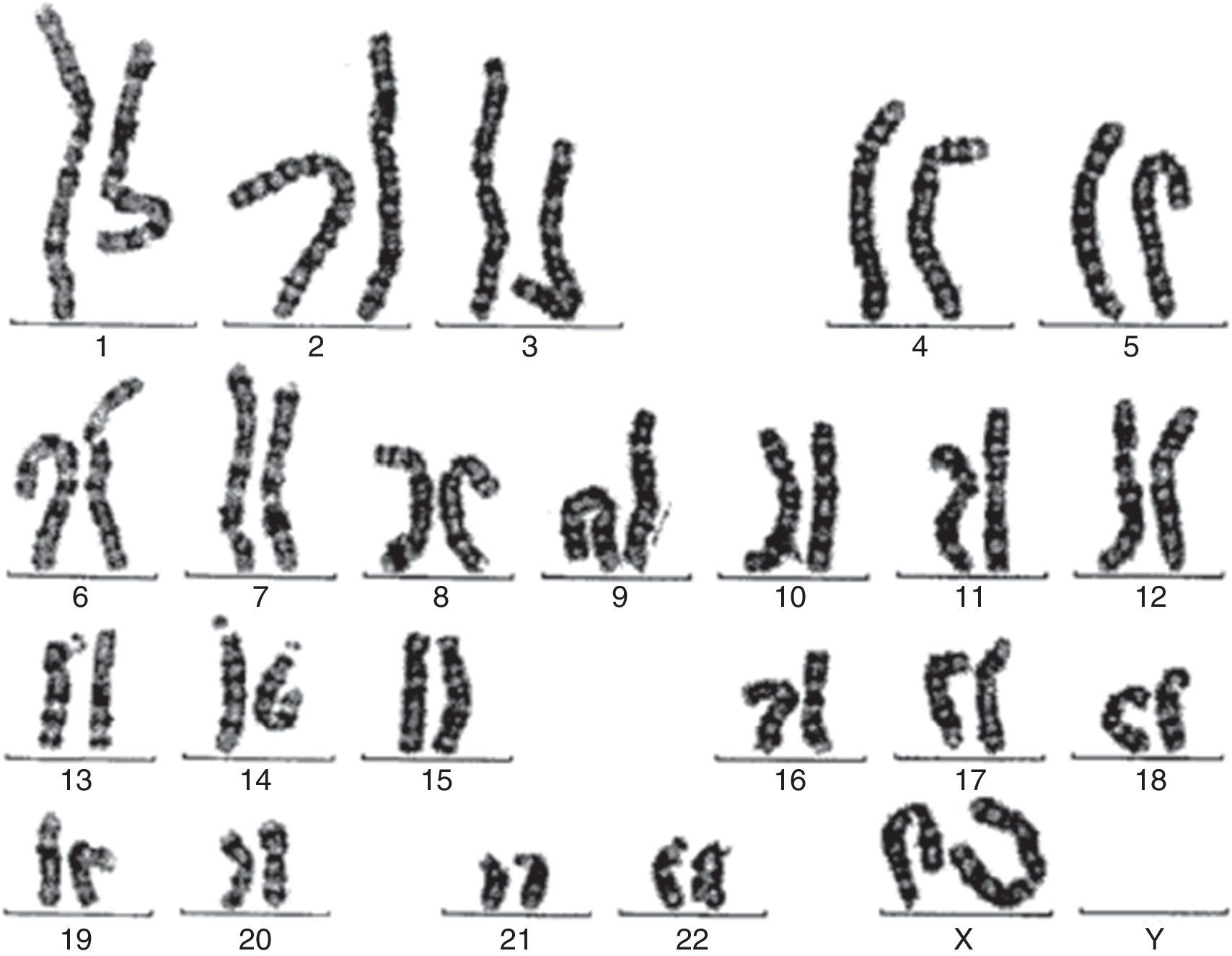
Se solicita cariotipo que muestra mediante técnica de bandas GTL 2 líneas celulares diferentes: un 20% de células 46,X con isocromosoma Xq, y el 80% restante 45,XO.
DiscusiónEl diagnóstico clínico del síndrome de Turner es muy variable en función de la etapa de la vida en la que se determine. En el período prenatal se puede observar higroma quístico nucal o generalizado, en el neonatal, linfedema y en la infancia, talla baja. En adolescentes se puede diagnosticar por retraso puberal o amenorrea primaria.
Es conocido que el síndrome de Turner ocasiona talla baja por pérdida de genes críticos relacionados con la talla presentes en el cromosoma X. La talla baja presente en el 100% de los casos es punto de partida del diagnóstico al ser en ocasiones el único síntoma. Sin embargo, la talla final suele guardar relación con la talla media parental, por lo que aquellos pacientes que se encuentren en percentiles superiores al p50 de la población serán quienes tengan mayor demora diagnóstica, especialmente si no se observan otras manifestaciones fenotípicas.
Algunas pacientes por tanto no serán diagnosticadas hasta el inicio de la pubertad, donde el potencial de crecimiento perdido será mayor, ya que aun no habrán recibido tratamiento con GH, privándoseles de la ganancia de talla final que conlleva.
Los factores que más influyen en los resultados de tratamiento con GH son la edad de inicio, la duración del tratamiento y la dosis recibida antes de la estrogenización, por lo que el diagnóstico precoz es un objetivo primordial del médico clínico.
El inicio de la terapia con hormona de crecimiento debe estar indicado en el momento en el que la talla de la paciente con síndrome de Turner se encuentra por debajo del percentil 5 con relación a su edad, lo que acontece habitualmente entre los 2-5 años2. Además, el inicio del tratamiento mejora la composición corporal, aumentando la masa magra y disminuyendo la masa grasa3.
De hecho, se ha demostrado que el inicio precoz de la terapia con hormona de crecimiento en niñas con síndrome de Turner mejora e incluso normaliza la talla final en algunos casos4–7. De igual forma, el inicio tardío de este tratamiento en pacientes con edades comprendidas entre los 10-13 años conlleva un aumento de talla de entre 2,1 y 7cm respecto a los controles no tratados8,9, por lo que el beneficio sigue siendo favorable aunque en menor cuantía.
Más de un tercio de los pacientes con síndrome de Turner tienen algún grado de mosaicismo10–12. La identificación genotípica del mismo depende directamente del método utilizado para su diagnóstico, variando desde un 34% con técnicas citogenéticas convencionales hasta un 60% con técnicas de hibridación in situ con fluorescencia13. Clásicamente se asocia la presencia de isocromosomas X con ausencia de anomalías congénitas, y con alta tasa de enfermedades autoinmunes (enfermedad de Crohn, tiroiditis linfocitaria) y sordera, lo que podría conllevar un retraso diagnóstico como en los casos presentados14,15. Las consecuencias fenotípicas de estos isocromosomas dependen de 2 factores: la naturaleza de los genes que se han perdido y el porcentaje de células 45,X; Asimismo, tampoco se encuentran prácticamente nunca formando mosaicismos con poblaciones XY normales, lo que traduce el hecho de que la anormalidad cromosómica tiene origen en la meiosis paternal16,17.
En cualquier caso, la talla baja del síndrome de Turner radica en la haploinsuficiencia del gen SHOX en el brazo corto del cromosoma X, por lo que cuando existen isocromosomas de los brazos largos del cromosoma X la talla baja es un hallazgo común a todos ellos18–20. Otros hallazgos fenotípicos relacionados con haploinsuficiencia genética en pacientes con isocromosoma Xq son la alteración de genes linfogénicos, y su manifestación craneofacial y cervical por compresión del sistema linfático. Otras manifestaciones en relación con esta alteración linfogénica serían la presencia de metacarpianos cortos, cúbito valgo o micrognatia, aunque se dan en menor proporción que en pacientes con monosomía X19,20.
Nuestras 2 pacientes muestran una talla genética de 163,2±5cm y 171,5±5cm, que se encuentran en percentiles medios-elevados de la población; por lo que el potencial de crecimiento perdido puede no ser tan obvio como en aquellos casos en los que la talla media paternal esté en percentiles inferiores. Además, ambas presentan escasa manifestación fenotípica del síndrome de Turner, por lo que todo ello pudo conducir a un retraso en la sospecha diagnóstica hasta edades más tardías.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
